Toxicidade dos Fármacos
NOÇÕES BÁSICAS EM FARMACOLOGIA
1 Toxicidade dos Fármacos:
À semelhança de muitas intervenções clínicas, o uso de fármacos para benefício terapêutico está sujeito à lei das consequências não pretendidas. Estas – denominadas efeitos colaterais, efeitos adversos ou efeitos tóxicos – constituem uma função dos mecanismos de ação dos fármacos, da magnitude da dose, das características e do estado de saúde do paciente. Assim, os princípios de farmacologia apresentados nos capítulos anteriores também se aplicam à farmacotoxicologia. Muitos capítulos subsequentes contêm tabelas de resumo farmacológico que listam, entre outras propriedades, os efeitos adversos específicos que podem ser causados por fármacos. O presente capítulo trata dos mecanismos subjacentes a esses efeitos.
De modo geral, os efeitos adversos incluem desde os comuns e relativamente benignos até os que representam sério risco de lesão orgânica ou morte. Entretanto, mesmo os primeiros podem causar considerável desconforto e levar o paciente a evitar o medicamento ou reduzir seu uso. Além disso, em geral, o tipo e o risco de efeitos adversos dependem da margem de segurança entre a dose necessária do fármaco para ser eficaz e a dose que provoca efeitos indesejáveis. Quando a margem de segurança é grande, a reação (efeito tóxico) resulta principalmente de superdosagem; quando pequena ou inexistente, os efeitos adversos podem manifestar-se com doses terapêuticas. Esses princípios aplicam-se também aos fármacos de venda não sujeita a prescrição, como o paracetamol e o ácido acetilsalicílico. Convém observar que as margens de segurança constituem uma função não apenas do fármaco, mas também do paciente, visto que a genética ou outras características – polimorfismos de enzimas que destoxificam metabólitos deletérios, comorbidades ou redução das reservas funcionais em órgão-chave – tornam os pacientes mais ou menos capazes de defender-se contra reações adversas. Este é um motivo pelo qual, sendo todas as outras variáveis iguais, novos medicamentos precisam ser iniciados nas menores doses provavelmente terapêuticas.
No início do desenvolvimento, os estudos pré-clínicos e clínicos são realizados para avaliar potência, seletividade, perfis farmacocinéticos e metabólicos e toxicidade dos compostos. Antes da comercialização, as agências reguladoras responsáveis pela aprovação do fármaco procedem a uma 1. 2. 3. revisão dos dados dos testes e decidem se os benefícios do medicamento superam seus riscos. Uma vez comercializado, e após exposição de inúmeros pacientes ao fármaco, o aparecimento de tipos ou frequências inesperados de efeitos adversos pode determinar reavaliação do medicamento, de modo que seu uso passe a ser restrito a grupos específicos de pacientes ou que seja retirado totalmente do mercado (como no caso do anti-inflamatório não esteroide rofecoxibe e do antidiabético troglitazona).
Mecanismos de toxicidade dos fármacos:
A possibilidade de um fármaco causar mais prejuízo do que benefício a determinado paciente depende de muitos fatores, incluindo idade, constituição genética, condições preexistentes, dose do fármaco administrado e outros fármacos já em uso. Por exemplo, indivíduos muito idosos ou crianças muito pequenas podem ser mais suscetíveis aos efeitos tóxicos, devido às diferenças dependentes da idade no perfil farmacocinético ou nas enzimas envolvidas no metabolismo dos fármacos. Conforme discutido no Capítulo 4, fatores genéticos podem determinar características individuais no metabolismo dos fármacos, na atividade dos receptores ou nos mecanismos de reparo. Pode haver maior tendência a reações medicamentosas adversas em pacientes com condições preexistentes, como disfunção hepática ou renal e, naturalmente, em pacientes alérgicos a fármacos específicos. Os medicamentos usados concomitantemente podem produzir confusão tanto na eficácia quanto na toxicidade, em especial quando compartilham ou modulam as mesmas vias metabólicas ou os mesmos transportadores. As interações medicamentosas com suplementos também constituem causa importante, porém frequentemente pouco reconhecida, de toxicidade dos fármacos. As interações medicamentosas e as interações entre fármacos e fitoterápicos serão discutidas, posteriormente, neste capítulo. A determinação clínica da toxicidade de um fármaco nem sempre pode ser direta: conforme observado no caso da Sra. G, por exemplo, é possível que um paciente tratado com antibiótico para combater infecção desenvolva febre alta, exantema cutâneo e morbidade significativa, devido à recidiva da infecção ou, em vez disso, a reação adversa ao antibiótico
Embora um espectro de efeitos adversos possa estar associado ao uso de qualquer fármaco ou classe de fármacos, é útil conceituar os mecanismos de farmacotoxicidade com base em vários paradigmas gerais:
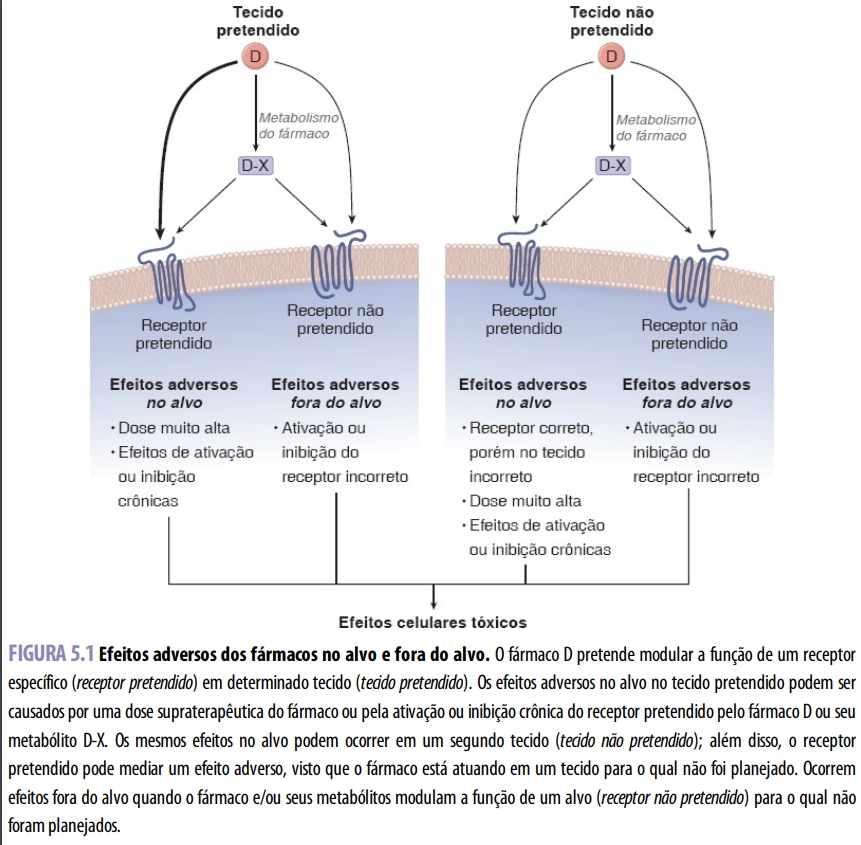
- Efeitos adversos no alvo, que resultam da ligação do fármaco a seu receptor pretendido, porém em concentração inapropriada, com cinética subótima ou no tecido incorreto
- Efeitos adversos fora do alvo, causados pela ligação do fármaco a um alvo ou receptor não pretendido
- Efeitos adversos mediados pelo sistema imune (Figura 5.2) Respostas idiossincrásicas cujo mecanismo não é conhecido.
Esses mecanismos são discutidos adiante. Convém observar que muitos fármacos podem ter efeitos direcionados para o alvo e fora dele, e que os efeitos adversos observados em pacientes podem ser causados por múltiplos mecanismos.
Efeitos no alvo:
Um conceito importante na toxicidade de substâncias é o fato de que um efeito adverso pode representar exagero da ação farmacológica desejada, devido a alterações na exposição à substância. Isso é possível em consequência de um erro deliberado ou acidental na dose, alterações na farmacocinética da substância (p. ex., em virtude de doença hepática ou renal ou de interações com outros fármacos) ou alterações na farmacodinâmica da interação fármaco-receptor, modificando a resposta farmacológica (mudanças na quantidade de receptores). Todas essas alterações podem acarretar aumento da concentração efetiva do fármaco e, portanto, aumento da resposta biológica. Como os efeitos no alvo são mediados pelo mecanismo da ação desejada do fármaco, são compartilhados com frequência por todos os membros da classe terapêutica; logo, são também conhecidos como efeitos de classe.
Um importante conjunto de efeitos adversos no alvo pode ocorrer em consequência da interação do fármaco ou de um de seus metabólitos com receptor apropriado, porém em tecidos diferentes daqueles afetados pela doença que está sendo tratada. Muitos alvos de fármacos são expressos em mais de um tipo celular ou tecidual. Por exemplo, o anti-histamínico difenidramina é um antagonista do receptor H1 utilizado para reduzir os efeitos da liberação de histamina em condições alérgicas. A difenidramina também atravessa a barreira hematencefálica e antagoniza os receptores H1 no sistema nervoso central, resultando em sonolência. Esse efeito adverso (colateral) levou ao desenvolvimento dos antagonistas dos receptores H1 de segunda geração, que não atravessam a barreira hematencefálica e que, portanto, não provocam sonolência. Notavelmente, o primeiro desses antagonistas, a terfenadina, produziu um efeito fora do alvo (interação com canais de potássio cardíacos) que acarretou efeito colateral diferente e grave – risco aumentado de morte cardíaca.
Os anestésicos locais, como lidocaína e bupivacaína, fornecem um segundo exemplo de efeito adverso no alvo. Eles foram desenvolvidos para impedir a transmissão de impulsos axônicos por meio do bloqueio dos canais de sódio nas membranas neuronais, próximo ao local de injeção. Esse bloqueio, seguido de superdosagem ou administração inapropriada (p. ex., administração intravascular), pode resultar em tremores, convulsões e morte. O haloperidol, agente antipsicótico, produz efeito benéfico mediante o bloqueio dos receptores D2 mesolímbicos e mesocorticais. Uma consequência do bloqueio desses receptores na hipófise consiste em aumento da secreção de prolactina, produzindo, em alguns casos, amenorreia, galactorreia, disfunção sexual e osteoporose.
Algumas vezes, os efeitos adversos no alvo revelam funções importantes do alvo biológico. Exemplo notável desse fenômeno é a administração de inibidores de hidroximetilgutaril coenzima A (HMG-CoA) redutase (as denominadas estatinas), utilizados clinicamente para diminuir os níveis de colesterol. O tecido-alvo pretendido desses fármacos é o fígado, onde inibem a HMG-CoA redutase, enzima que limita a velocidade na síntese de isoprenoides. Um efeito adverso raro do tratamento com estatinas é a toxicidade muscular, incluindo rabdomiólise e miosite. Tal efeito ressalta o papel fisiológico da HMG-CoA redutase na regulação da modificação pós-translacional de várias proteínas musculares por meio de um processo que envolve lipídios, denominado geranilgeranilação. As estatinas, como exemplo de fármacos que provocam lesão do músculo esquelético, também serão discutidas mais adiante neste capítulo.
Efeitos fora do alvo:
Ocorrem quando um fármaco interage com alvos não pretendidos. Alguns fármacos, de fato, são tão seletivos que interagem com um único alvo molecular. Exemplo notável desse tipo de efeito é fornecido pela interação de numerosos compostos com canais de potássio IKr cardíacos. (Como o gene humano relacionado ether-à-go-go [hERG] codifica uma unidade do canal IKr , esses canais são também denominados canais hERG.) A inibição das correntes de potássio transportadas pelos canais IKr pode levar a repolarização tardia dos miócitos cardíacos. Por sua vez, tal repolarização pode resultar em aumentos de frequência cardíaca corrigida por intervalo QT (QTc), arritmias cardíacas, incluindo torsade de pointes, e morte súbita. O anti-histamínico terfenadina foi um dos primeiros exemplos de compostos que interferem nas correntes dos canais de potássio cardíacos, produzindo arritmias potencialmente fatais. Esse fármaco foi planejado para evitar a sonolência, efeito adverso dos antagonistas dos receptores H1 de primeira geração (ver discussão anterior). A observação de aumento na incidência de mortes por arritmias cardíacas em pacientes usando terfenadina ocasionou sua retirada do mercado e induziu esforços vigorosos para compreender como impedir esses eventos. Os levantamentos realizados mostraram que, embora muitos compostos inibam o canal hERG, aqueles com metade da concentração inibitória máxima (IC50) de mais de 30 vezes a concentração plasmática na dose terapêutica recomendada (Cmáx. , ajustada para a ligação às proteínas) apresentam baixo risco de causar prolongamento do QTc e arritmias cardíacas. Posteriormente, foi descoberto que o metabólito ativo da terfenadina, a fexofenadina, inibe apenas de maneira fraca o canal de hERG, de modo que, hoje em dia, a fexofenadina é comercializada como anti-histamínico mais seguro.
Como numerosos compostos têm a capacidade de interferir nos canais de potássio cardíacos, todos os novos candidatos a fármacos são avaliados quanto a seu potencial de interagir com esses canais promíscuos. Em ensaio do hERG, o efeito potencial de compostos sobre as correntes de potássio cardíacas em seres humanos é medido em um sistema in vitro que utiliza células transfectadas com o gene humano relacionado ether-à-go-go (hERG). Além do ensaio do hERG, o potencial de alteração da eletrofisiologia cardíaca é avaliado em um modelo animal não roedor. Como requisito para a aprovação de sua comercialização, os novos fármacos também são avaliados clinicamente quanto à sua capacidade de prolongar o QTc em seres humanos. Os compostos que aumentam o QTc acima de um valor determinado com exposição próxima àquela necessária para produzir o efeito terapêutico são considerados como tendo algum risco de produzir arritmias. O controle positivo usado na maioria desses estudos “completos” do QTc é, principalmente, o moxifloxacino, antibiótico que aumenta o QTc em doses clínicas, mas que está associado a baixo risco de arritmogênese.
Os enantiômeros (isômeros especulares) de um fármaco também podem produzir efeitos fora do alvo. Conforme descrito no Capítulo 1, os receptores de fármacos são, em especial, sensíveis ao arranjo tridimensional dos átomos na molécula do fármaco; por conseguinte, frequentemente são capazes de diferenciar os enantiômeros de um fármaco. Exemplo trágico e bem conhecido desse fenômeno ocorreu com a administração da talidomida racêmica (mistura dos enantiômeros [R] e [S]) na década de 1960 para tratamento do enjoo matinal em gestantes. Enquanto o enantiômero (R) da talidomida era um sedativo eficaz, o enantiômero (S) era um potente teratógeno, que provocou graves defeitos congênitos, como amelia (ausência de membros) e vários graus de focomelia em número estimado de 10.000 recém-nascidos em 46 países (mas não nos EUA, graças a Frances Kelsey da Food and Drug Administration [FDA], que duvidou da segurança da talidomida).
A probabilidade de diferenças farmacológicas significativas entre enantiômeros levou a FDA a avaliar esses compostos como entidades clínicas separadas. Quando é possível demonstrar que uma única preparação enantiomérica de um fármaco apresenta melhores propriedades farmacológicas do que uma versão racêmica, o enantiômero purificado pode ser aprovado como novo fármaco. Por exemplo, o inibidor racêmico da bomba de prótons, o omeprazol, e seu enantiômero (S), o esomeprazol ([S]-omeprazol), são comercializados como fármacos separados. Outro efeito comum fora do alvo é a ativação não pretendida de diferentes subtipos de receptores. Por exemplo, o receptor β1-adrenérgico é expresso no coração, e sua ativação aumenta a frequência cardíaca e a contratividade miocárdica. Receptores β2-adrenérgicos estreitamente relacionados são expressos principalmente em células musculares lisas das vias respiratórias e da vasculatura, e a ativação desses receptores β2 provoca relaxamento do músculo liso e dilatação desses tecidos (Capítulo 10). Os usos clínicos dos antagonistas dos receptores β-adrenérgicos (os denominados βbloqueadores) são frequentemente direcionados para o receptor β1 , a fim de controlar a frequência cardíaca e reduzir a demanda de oxigênio do miocárdio em pacientes com angina ou insuficiência cardíaca. Todavia, alguns antagonistas desses receptores não são totalmente seletivos e também podem antagonizar o receptor β2 . Sendo assim, os antagonistas não seletivos dos receptores βadrenérgicos estão contraindicados para pacientes com asma, visto que esses fármacos podem causar inadvertidamente broncoconstrição por meio de antagonismo dos receptores β2 . De modo semelhante, o uso de agonistas β2 inalados no tratamento da asma, particularmente em altas doses, pode produzir aumento da frequência cardíaca.
O segundo efeito fora do alvo devido à ativação não pretendida de diferentes subtipos de receptores é a valvopatia causada pelo agente anorético fenfluramina. O principal mecanismo de ação desse fármaco parece envolver liberação de serotonina (5-hidroxitriptamina [5-HT]) e inibição da recaptação da 5-HT em áreas do cérebro que regulam o comportamento alimentar. Entretanto, o composto também ativa os receptores 5-HT2B, induzindo proliferação de miofibroblastos nas valvas atrioventriculares. Além disso, pode ocorrer desenvolvimento de hipertensão pulmonar, ocasionando, em alguns casos, a morte. Em virtude desse efeito adverso, a fenfluramina foi retirada do mercado (ver Toxicidade cardiovascular induzida por fármacos). Os potenciais efeitos fora do alvo de alguns fármacos podem ser explorados em camundongos ou ratos de laboratório geneticamente modificados, nos quais o receptor-alvo pretendido sofre deleção (às vezes apenas em tecidos específicos). Se o fármaco afetar de alguma maneira esses roedores, outros alvos além do pretendido devem estar envolvidos.
Os efeitos fora do alvo de determinados fármacos e seus metabólicos podem ser estabelecidos apenas empiricamente, ressaltando a importância de testes extensos tanto em experimentos préclínicos quanto em estudos clínicos. Apesar desses testes, certas toxicidades raras dos fármacos são descobertas apenas quando se dá exposição em população muito maior do que a exigida para estudos clínicos. Por exemplo, as fluoroquinolonas, classe de antibióticos de amplo espectro derivados do ácido nalidíxico, apresentam toxicidade mínima nos estudos pré-clínicos e ensaios clínicos. Entretanto, o uso clínico mais amplo desses fármacos levou a relatos de anafilaxia, prolongamento do intervalo QTc e cardiotoxicidade potencial, tendo como consequência a retirada do mercado de dois fármacos dessa classe, temafloxacino e grepafloxacino. O uso de outra fluoroquinolona, trovafloxacino, está significativamente restrito por causa de sua hepatotoxicidade imprevista. Em comparação, ciprofloxacino e levofloxacino são, em geral, bem tolerados e utilizados com frequência no tratamento de infecções bacterianas. Contudo, conforme observado no caso descrito na introdução do capítulo, até mesmo esses agentes podem ocasionalmente provocar grave reação de hipersensibilidade.
Toxicidade idiossincrásica:
Reações medicamentosas idiossincrásicas são efeitos adversos que aparecem de modo imprevisível em fração muito pequena de pacientes, por motivos desconhecidos. Esses efeitos não se manifestam nos testes realizados antes da comercialização, seja em animais de laboratório, seja em pacientes. O estudo sistemático de variações nas respostas dos pacientes a diferentes fármacos pode ajudar a elucidar a genética ou outros mecanismos subjacentes às reações medicamentosas idiossincrásicas. O aparecimento de lesão idiossincrásica produzindo disfunção orgânica permanente e/ou morte, mesmo quando rara, por vezes determina a retirada do fármaco do mercado, precisamente porque não é possível identificar populações de pacientes suscetíveis.
2 Contextos da toxicidade dos fármacos:
Superdose de fármacos:
O médico e alquimista suíço Paracelsus assinalou, há quase 500 anos, que “todas as substâncias são venenos; não existe nenhuma que não seja veneno. A dose correta é que diferencia um veneno de um remédio”. Em alguns casos, como na tentativa de suicídio, a superdose de um fármaco é intencional. Entretanto, a maioria dos casos de superdose ocorre de modo acidental. Estima-se que eventos adversos de fármacos em decorrência de erros de dosagem afetem cerca de 775.000 pessoas por ano, com custo hospitalar anual associado de 1,5 a 5,5 bilhões de dólares. Esse custo significativo tanto para o paciente quanto para o sistema de assistência à saúde motivou esforços sistemáticos para minimizar erros na prescrição e nas práticas de dosagem.
Interações medicamentosas:
À medida que a população envelhece, e múltiplos medicamentos são prescritos a um número crescente de pacientes, o potencial de interações medicamentosas aumenta. Foram identificadas numerosas interações adversas, cujos mecanismos frequentemente envolvem efeitos farmacocinéticos ou farmacodinâmicos. As interações entre fármacos e fitoterápicos também constituem importante subgrupo de interações medicamentosas.
Interações medicamentosas farmacocinéticas:
As interações farmacocinéticas surgem quando um fármaco modifica absorção, distribuição, metabolismo ou excreção de outro fármaco, alterando, assim, a concentração desse fármaco ativo no organismo. Conforme discutido no Capítulo 4, alguns fármacos podem inibir ou induzir as enzimas hepáticas do citocromo P450. Quando dois fármacos são metabolizados pela mesma enzima P450, a inibição competitiva ou irreversível dessa enzima pode provocar aumento na concentração plasmática do segundo fármaco. Por outro lado, a indução de uma enzima P450 específica por um fármaco pode resultar em diminuição das concentrações plasmáticas de outros fármacos metabolizados pela mesma enzima. O antifúngico cetoconazol é um potente inibidor da enzima 3A4 do citocromo P450 (CYP3A4). A coadministração de fármacos também metabolizados pela CYP3A4 pode produzir diminuição do metabolismo desses fármacos e aumento de seus níveis plasmáticos. Se o fármaco coadministrado tiver baixo índice terapêutico, é possível ocorrer toxicidade. Em virtude da potente inibição da CYP3A4, o cetoconazol é frequentemente utilizado em estudos clínicos planejados para avaliar a importância das interações medicamentosas farmacocinéticas.
Além de alterar a atividade das enzimas P450, os fármacos podem afetar o transporte de outros fármacos para dentro e para fora dos tecidos. Segundo o que foi abordado no Capítulo 4, a Pglicoproteína (Pgp), codificada pelo gene de resistência a múltiplos fármacos 1 (MDR1), é uma bomba de efluxo que transporta fármacos para o lúmen intestinal. A administração de um fármaco que a inibe ou que é substrato dela pode acarretar aumento nas concentrações plasmáticas de outros fármacos normalmente bombeados para fora do corpo por esse mecanismo. Como a Pgp também atua no transporte de fármacos através da barreira hematencefálica, os compostos que a inibem podem afetar o transporte de fármacos no SNC. Outros transportadores, como o polipeptídio transportador de ânions orgânicos 1 (OATP1), medeiam a captação de fármacos nos hepatócitos para seu metabolismo, bem como o transporte de fármacos através do epitélio tubular dos rins para excreção; ambos os mecanismos promovem a depuração do fármaco do corpo. As interações de um fármaco ou de um de seus metabólitos com essas classes de transportadores podem ocasionar concentrações plasmáticas inapropriadamente altas de outros fármacos processados pelo mesmo transportador.
Às vezes, uma interação farmacocinética pode ser desejável. Por exemplo, como a penicilina é depurada por secreção tubular nos rins, a meia-vida de eliminação desse fármaco pode ser aumentada se for administrado de modo concomitante com probenecida, inibidor do transporte tubular renal. Um segundo exemplo é fornecido pela combinação do imipeném, antibiótico de amplo espectro, com a cilastatina, inibidor seletivo de uma dipeptidase da borda em escova renal (desidropeptidase I). Como o imipeném é rapidamente inativado pela desidropeptidase I, a coadministração dele com cilastatina é usada para obter concentrações plasmáticas terapêuticas do antibiótico. Um fármaco que se liga às proteínas plasmáticas, como a albumina, pode deslocar um segundo fármaco da mesma proteína, aumentando sua concentração plasmática livre e, consequentemente, sua biodisponibilidade para tecidos-alvo e tecidos não alvo. Esse efeito pode ser intensificado em uma situação na qual os níveis circulantes de albumina estão baixos, como em insuficiência hepática, desnutrição (síntese diminuída de albumina) ou síndrome nefrótica (excreção aumentada de albumina).
Interações medicamentosas farmacodinâmicas:
As interações farmacodinâmicas surgem quando um fármaco modifica a resposta dos tecidos-alvo ou não alvo a outro fármaco. Podem ocorrer interações farmacodinâmicas tóxicas quando dois fármacos ativam vias complementares, resultando em efeito biológico exagerado. Esse tipo de interação é observado com a coadministração de sildenafila (para disfunção erétil) e nitroglicerina (para angina de peito). A sildenafila inibe a fosfodiesterase tipo 5 (PDE5) e, portanto, prolonga a ação do GMP cíclico (GMPc), enquanto a nitroglicerina estimula a guanilil ciclase a aumentar os níveis de GMPc no músculo liso vascular.
Um segundo exemplo consiste na coadministração de agentes antitrombóticos. Após cirurgia de substituição de quadril, os pacientes são tratados com varfarina profilática durante várias semanas para impedir o desenvolvimento de trombose venosa profunda no pós-operatório. Como as concentrações plasmáticas de varfarina podem não alcançar um nível terapêutico durante vários dias, algumas vezes a heparina de baixo peso molecular e a varfarina são administradas concomitantemente durante esse período. Entretanto, conforme observado no caso da Sra. G, pode ocorrer sangramento significativo se os efeitos da heparina e da varfarina forem sinérgicos, produzindo níveis supraterapêuticos de anticoagulação.
Interações entre fármacos e fitoterápicos:
A segurança e a eficácia de um fármaco também podem ser alteradas pela coexposição a vários produtos não farmacêuticos, como alimentos, bebidas, fitoterápicos ou suplementos dietéticos. Muitos fitoterápicos consistem em misturas complexas de compostos biologicamente ativos, e sua segurança e efetividade raramente foram testadas em estudos controlados. O largo uso de fitoterápicos não regulamentados entre o público deve conduzir o médico a investigar o uso desses produtos pelo paciente. A literatura apresenta diversos relatos de falha terapêutica de fármacos tomados com fitoterápicos, assim como alguns que apontam toxicidade. Por exemplo, a preparação ginkgo biloba (da árvore do mesmo nome) inibe a agregação plaquetária. O uso concomitante de ginkgo e de antiinflamatórios não esteroides (AINE), que também inibem a agregação plaquetária, pode aumentar o risco de sangramento. Os produtos de Echinacea contêm alcaloides que podem causar depleção das reservas hepáticas de glutationa, aumentando o risco de toxicidade do paracetamol. Em combinação com inibidores seletivos da recaptação da serotonina, a erva-de-são-joão pode causar síndrome serotoninérgica leve.
Mecanismos celulares de toxicidade: apoptose e necrose:
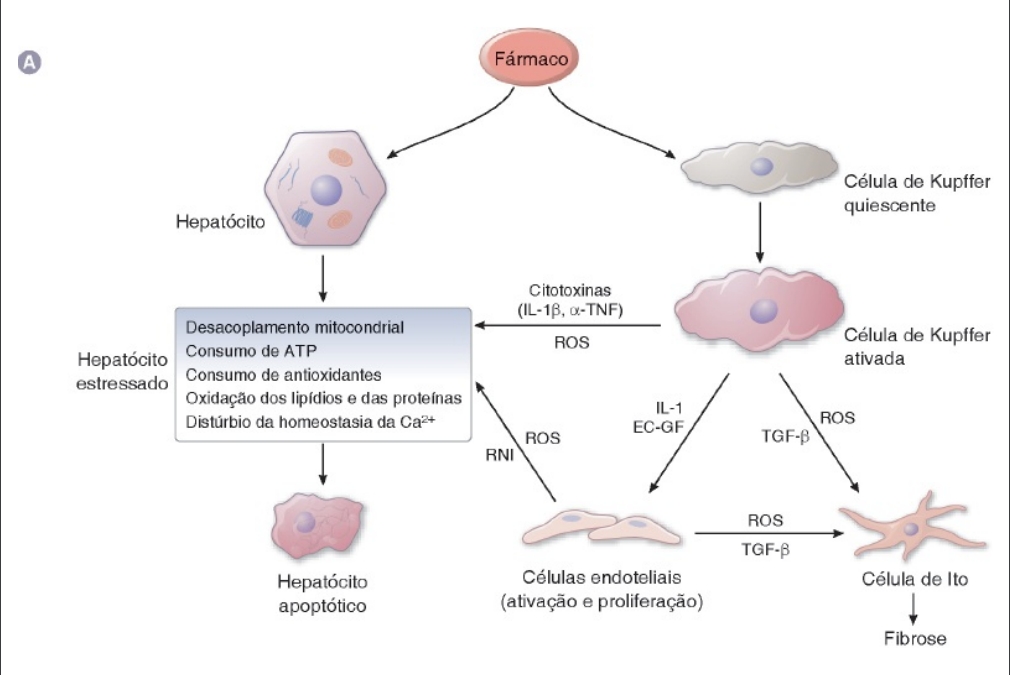
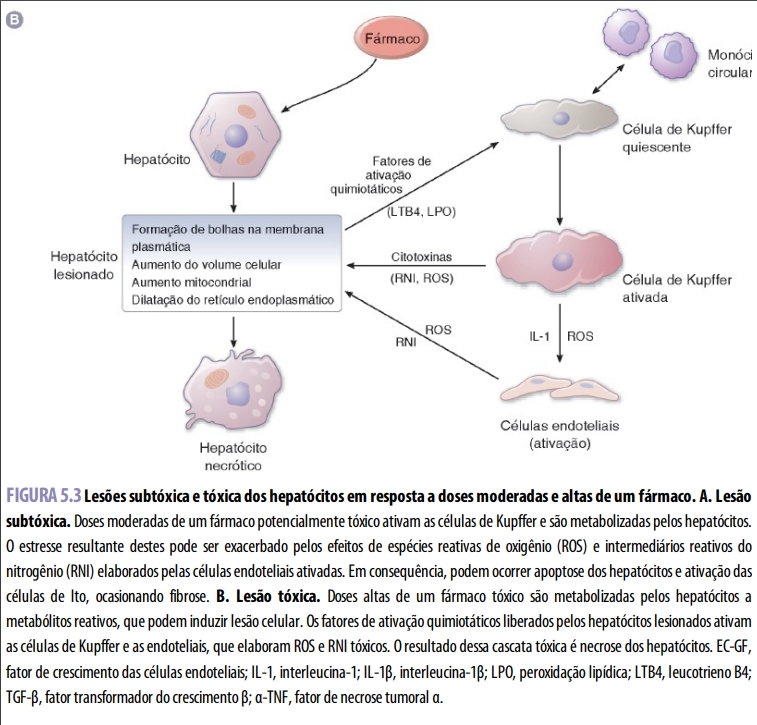
As células dispõem de vários mecanismos para evitar ou reparar lesões, e ocorre toxicidade quando essas defesas são sobrepujadas. Em alguns casos, a toxicidade pode ser minimizada a curto prazo, porém agressões repetidas (p. ex., as que levam à fibrose) finalmente são capazes de comprometer a função dos órgãos. As respostas celulares primárias a um fármaco potencialmente tóxico estão ilustradas na Figura 5.3A, B, usando o hepatócito como exemplo. Dependendo da gravidade da agressão tóxica, uma célula pode sofrer apoptose (morte celular programada) ou necrose (morte celular não controlada). No processo de apoptose, a célula sofre autodestruição ordenada pela atividade coordenada de diversas proteínas específicas. A apoptose pode ser benéfica quando elimina células lesionadas sem lesão do tecido circundante. A inibição desse processo é comum em muitas células cancerosas. Se a agressão tóxica for tão grave a ponto de impedir a morte celular ordenada, a célula sofre necrose. Esta se caracteriza por digestão enzimática do conteúdo celular, desnaturação das proteínas celulares e ruptura das membranas. Enquanto as células apoptóticas sofrem morte celular com inflamação e ruptura mínimas do tecido adjacente, as células necróticas atraem células inflamatórias e podem causar lesão das células adjacentes sadias.
Toxicidade dos órgãos e tecidos:
A maioria dos capítulos deste livro contém tabelas que apresentam efeitos adversos graves e comuns dos fármacos discutidos no capítulo. Aqui, consideram-se os mecanismos gerais de lesão e reparo relacionados com efeitos tóxicos dos fármacos nos principais sistemas orgânicos. Este capítulo não pretende catalogar todas as possíveis lesões de cada órgão ou sistema orgânico, visto que a gama de toxicidade orgânica e tecidual associada a fármacos é tão grande que torna impossível discutir todas as toxicidades específicas de todos os fármacos em um único capítulo. Por esse motivo, são apresentados alguns exemplos específicos de lesão para demonstrar as características gerais da farmacotoxicidade.
Respostas imunes deletérias e imunotoxicidade:
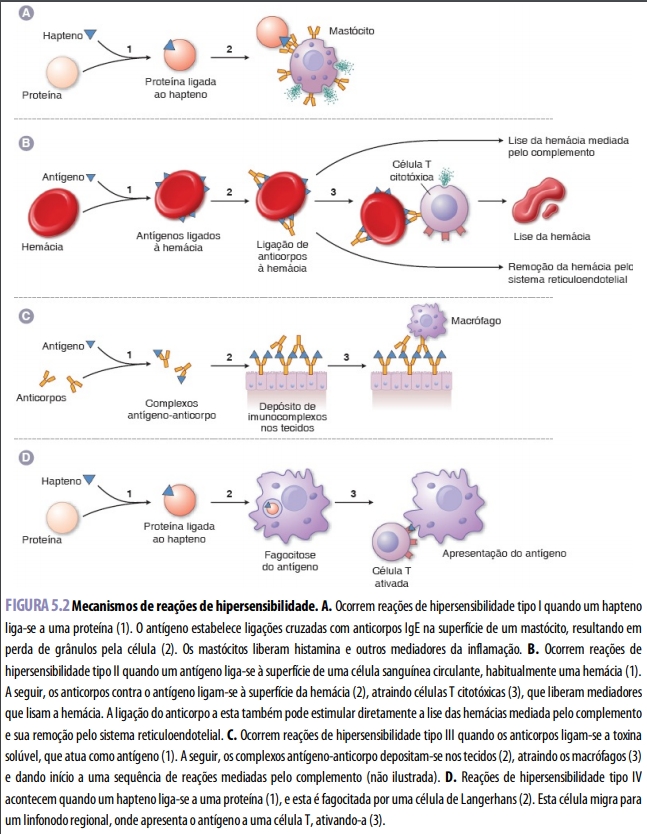
A estimulação do sistema imune desempenha um papel na toxicidade de vários fármacos e classes de fármacos. Estes podem ser responsáveis por reações imunes (reações clássicas de tipo I até tipo IV), síndromes que simulam algumas características das respostas imunes (síndrome do homem vermelho) e exantemas cutâneos (erupções), incluindo afecções graves e potencialmente fatais, como síndrome de Stevens-Johnson e necrólise epidérmica tóxica. Os fármacos também podem comprometer a função normal do sistema imune (imunotoxicidade), resultando em efeitos secundários, como risco aumentado de infecção. É possível que alguns fármacos sejam reconhecidos pelo sistema imune como substâncias estranhas. Os que consistem em pequenas moléculas, com massa inferior a 600 dáltons, não são imunogênicos diretos, mas podem atuar como haptenos, em que o fármaco liga-se (por vezes, de modo covalente) a uma proteína no corpo e, em seguida, torna-se capaz de deflagrar uma resposta imune. Se um fármaco for grande o suficiente (p. ex., um peptídio ou uma proteína terapêuticos), será capaz de ativar diretamente o sistema imune. Os dois mecanismos imunes principais pelos quais os fármacos podem provocar lesão são as respostas de hipersensibilidade (respostas alérgicas) e as reações autoimunes.
As respostas de hipersensibilidade são classicamente divididas em quatro tipos, cujos mediadores e manifestações clínicas constam na Tabela 5.1. É necessária exposição prévia a uma substância para a ocorrência de cada tipo de reação.
A resposta de hipersensibilidade tipo I (hipersensibilidade imediata ou anafilaxia) resulta da produção de IgE após exposição a um antígeno, que pode ser uma proteína estranha, como o agente trombolítico derivado de bactéria, a estreptoquinase, ou uma proteína endógena modificada por um hapteno para se tornar imunogênica. Fragmentos de penicilina – na formulação a ser administrada ou formados in vivo – podem atuar como haptenos e ativar o sistema imune. A exposição subsequente ao antígeno provoca exocitose dos grânulos dos mastócitos, com liberação de mediadores inflamatórios, como histamina e leucotrienos, que promovem broncoconstrição, vasodilatação e inflamação. As respostas de hipersensibilidade tipo I manifestam-se, na pele, como reação de pápula e eritema. Podem surgir sintomas de “febre do feno”, como conjuntivite e rinite, nas vias respiratórias superiores, e broncoconstrição asmática nas vias respiratórias inferiores (Capítulo 47). A resposta de hipersensibilidade tipo II (hipersensibilidade citotóxica dependente de anticorpos) ocorre quando um fármaco liga-se a células, habitualmente hemácias, e é reconhecido por um anticorpo, em geral IgG. Este desencadeia a lise da célula por fixação do complemento, fagocitose pelos macrófagos ou citólise por células T citotóxicas. As respostas adversas tipo II são raras, mas comuns a diversos fármacos, incluindo penicilina e quinidina.
Respostas de hipersensibilidade tipo III (hipersensibilidade mediada por imunocomplexos) manifestam-se quando há formação de anticorpos, normalmente IgG ou IgM, contra antígenos solúveis. Os complexos antígeno-anticorpo depositam-se em tecidos, como rins, articulações e endotélio vascular pulmonar, provocando lesão e iniciando uma resposta inflamatória, a doença do soro, em que ocorre ativação dos leucócitos e do complemento dos tecidos. Por exemplo, a hipersensibilidade tipo III pode ser causada pela administração de antivenenos, isto é, proteínas séricas equinas obtidas pela inoculação, em um cavalo, do veneno a ser neutralizado. Exemplos de outros fármacos que podem estar associados a risco de doença do soro são bupropiona e cefaclor. A resposta de hipersensibilidade tipo IV (hipersensibilidade de tipo tardio) resulta da ativação das células TH1 e T citotóxicas. Com mais frequência, manifesta-se como dermatite de contato, quando uma substância atua como hapteno e liga-se a proteínas do hospedeiro. A primeira exposição normalmente não produz resposta, mas exposições dérmicas subsequentes podem ativar as células de Langerhans que migram para os linfonodos locais e ativam as células T. Em seguida, estas retornam à pele e desencadeiam uma resposta imune. As respostas mais conhecidas de hipersensibilidade tipo IV incluem reações ao contato com a planta Toxicodendron radicans e desenvolvimento de alergia ao látex. A exposição repetida a um agente reconhecido pelo sistema imune como substância estranha pode desencadear resposta imune maciça. Essa “tempestade de citocinas” pode resultar em febre, hipotensão e até mesmo falência de órgãos. Por conseguinte, os médicos devem considerar a possibilidade de reações imunes a todos os fármacos administrados, inclusive àqueles que se mostraram seguros em populações mais amplas. No caso apresentado no início deste capítulo, a Sra. G teve febre e exantema provavelmente causados por uma reação de hipersensibilidade mediada pelas células T ao ciprofloxacino. Uma vez identificado o problema, e com a interrupção do ciprofloxacino, cessaram a febre e o exantema.
Ocorre autoimunidade quando o sistema imune do organismo ataca suas próprias células. Diversos fármacos e várias outras substâncias químicas podem desencadear esse tipo de reação. A metildopa pode causar anemia hemolítica ao deflagrar reação autoimune contra os antígenos rhesus (fatores Rh). Vários outros fármacos, como hidralazina, isoniazida e procainamida, podem causar síndrome semelhante ao lúpus ao induzir a produção de anticorpos dirigidos contra a mieloperoxidase (hidralazina e isoniazida) ou o DNA (procainamida). A síndrome do homem vermelho é observada em pequena porcentagem de pacientes aos quais se administram fármacos intravenosos, como o antibiótico vancomicina. A reação é causada pelo efeito direto desses fármacos sobre os mastócitos, desencadeando a ruptura de grânulos nessas células. Diferentemente das reações tipo I, essa ação na síndrome do homem vermelho é independente da presença de IgE pré-formada ou complemento. Tal síndrome associa-se ao aparecimento de pápulas cutâneas e urticária (semelhantes àquelas observadas nas reações tipo I); entretanto, trata-se, com frequência, de um fenômeno relativamente localizado, que acomete pescoço, braços e parte superior do tronco. Raramente essa síndrome evolui para toxicidade grave, como angioedema e hipotensão. Ela também tem sido denominada reação anafilactoide, em virtude de sua semelhança com a anafilaxia (reação tipo I). Como a síndrome do homem vermelho inicia-se pela ação direta de um fármaco sobre os mastócitos, surge tipicamente durante a infusão (p. ex., infusões de vancomicina são frequentemente administradas por um período de 60 min). A síndrome em geral tem sua gravidade diminuída ou desaparece após reduzir a velocidade de infusão ou suspendê-la. Também pode ser reduzida com o uso profilático de anti-histamínicos, e sua gravidade pode diminuir com administrações intravenosas repetidas. Além de vancomicina, ciprofloxacino, anfotericina B, rifampicina e teicoplanina podem causar essa reação. A síndrome do homem vermelho também se associa a determinados excipientes usados em formulações intravenosas, como Cremophor, excipiente para paclitaxel e ciclosporina.
É possível ocorrerem exantemas cutâneos após a administração de diversos fármacos; em geral esses exantemas são diagnosticados como eritema multiforme. As condições mais graves (algumas vezes potencialmente fatais), conhecidas como síndrome de Stevens-Johnson e necrólise epidérmica tóxica, foram relatadas com o uso de barbitúricos, sulfonamidas, antiepilépticos (fenitoína, carbamazepina), agentes anti-inflamatórios não esteroides (ibuprofeno, celecoxibe, valdecoxibe), alopurinol e outros fármacos. A patogenia da síndrome de Stevens-Johnson não está totalmente elucidada, porém a aparência morfológica da inflamação das mucosas e da pele, com desenvolvimento de bolhas e separação da epiderme da derme, é compatível com uma etiologia imune. Pode existir relação temporal entre a administração de um fármaco e o desenvolvimento de lesões cutâneas, porém alguns casos de síndrome de Stevens-Johnson são idiopáticos ou relacionados com infecção. Por esse motivo, nem todos os casos dessa síndrome podem ser atribuídos à exposição a um fármaco.
A imunotoxicidade ou lesão do sistema imune pode ocorrer como efeito adverso de tratamento farmacológico ou como propósito específico de terapia. Os agentes citotóxicos usados na quimioterapia do câncer são planejados para matar as células neoplásicas em proliferação, porém também provocam lesão das células normais em proliferação na medula óssea, nos tecidos linfoides, no intestino e nos folículos pilosos em concentrações do fármaco necessárias para sua eficácia. No caso desses agentes, existe, em geral, pouca margem de segurança para o prejuízo dos tecidos normais, e o tratamento bem-sucedido depende de maior sensibilidade das células cancerosas em comparação com os tecidos normais. Com frequência, a terapia com agentes citotóxicos para os leucócitos é acompanhada de risco aumentado de infecção. Pode-se elevar a margem entre efeitos adversos e efeitos terapêuticos com o uso de fármacos que estimulam a produção de leucócitos (filgrastim).
Talvez seja apropriado direcionar o tratamento para o sistema imune quando a doença é exacerbada por alguma resposta imune deletéria (Capítulo 45). Por exemplo, corticosteroides inalados podem ser prescritos para controlar os sintomas em pacientes que apresentam exacerbações graves e frequentes de doença pulmonar obstrutiva crônica (Capítulo 47). Entretanto, com a inibição das respostas imunes a microrganismos patogênicos, esse tratamento também está associado a um risco aumentado de pneumonia. Algumas imunoterapias são direcionadas para tipos específicos de células no sistema imune e associam-se a risco aumentado de infecções graves. O rituximabe é um anticorpo monoclonal (mAb) direcionado contra as células B (CD20-positivas), envolvidas na patogenia do linfoma não Hodgkin (células B CD20-positivas malignas) e da artrite reumatoide (células B CD-20-positivas produtoras de anticorpos). Foram observados dois efeitos adversos potencialmente graves com o uso do rituximabe: a leucoencefalopatia multifocal progressiva (LMP), infecção causada por um poliomavírus, o vírus JC (JCV), e a reativação da hepatite B com potencial de hepatite fulminante. Esses agentes infecciosos geralmente se encontram em forma latente nos pacientes antes do tratamento com rituximabe, porém a perda da imunocompetência em decorrência do tratamento possibilita a expressão dessas infecções graves. De modo semelhante, o efalizumabe é um anticorpo monoclonal cujo alvo é a CD11a, a subunidade α do antígeno 1 associado à função leucocitária (LFA-1), expressa em todos os leucócitos. O efalizumabe, ao diminuir a expressão da CD11a na superfície celular e inibir a ligação da LFA-1 à molécula de adesão intercelular 1 (ICAM-1), impede a adesão dos leucócitos e atua como imunoterapia efetiva para a psoríase.
Entretanto, como a CD11a também é expressa na superfície de células B, monócitos, neutrófilos, células natural killer e outros leucócitos, o efalizumabe pode afetar inclusive a ativação, a adesão, a migração e a destruição dessas células. À semelhança do rituximabe, o efalizumabe tem sido associado à LMP; esse efeito adverso grave provocou sua retirada do mercado em 2009. Foi observado aumento semelhante na frequência de LMP em pacientes tratados com natalizumabe para esclerose múltipla. Esse fármaco liga-se à subunidade α4 das integrinas α4β1 e α4β7 expressas na superfície de todos os leucócitos, exceto os neutrófilos; ao inibir a adesão dos leucócitos mediada pela subunidade α4 às células-alvo, o fármaco impede qualquer recrutamento e ativação adicionais dos leucócitos.
Hepatotoxicidade induzida por fármacos:
Conforme descrito no Capítulo 4, muitos fármacos são metabolizados no fígado, e alguns desses metabólitos podem causar lesão hepática. Exemplo clinicamente significativo é o paracetamol, amplamente usado como analgésico e antipirético. Em sua faixa posológica terapêutica, ele é metabolizado predominantemente por glicuronidação e sulfatação, resultando em metabólitos prontamente excretados; uma pequena fração da dose também é excretada em sua forma inalterada. Entretanto, como mostra a Figura 5.4, o paracetamol também pode ser oxidado a uma espécie reativa e potencialmente tóxica, a N-acetil-p-benzoquinoneimina (NAPQI). A glutationa pode conjugar-se com a NAPQI e, assim, destoxificá-la, porém a superdosagem de paracetamol provoca depleção das reservas de glutationa (o que também pode ocorrer em outras condições), deixando a NAPQI livre para atacar proteínas celulares e mitocondriais, resultando finalmente em necrose dos hepatócitos. A administração do antídoto N-acetilcisteína (NAC) no momento apropriado (cerca de 10 h após a superdosagem de paracetamol) produz a restauração das reservas de glutationa, podendo impedir a ocorrência de insuficiência hepática e morte. Esse exemplo ressalta a importância da dose: embora o paracetamol seja usado com segurança por milhões de indivíduos diariamente, o mesmo fármaco, quando tomado em excesso, é responsável por cerca de 50% dos casos de insuficiência hepática aguda nos EUA.
A hepatotoxicidade inesperada constitui o motivo mais frequente das retiradas de fármacos do mercado nos EUA. Como muitos casos de hepatite fulminantes após terapia farmacológica são idiossincrásicos – isto é, o mecanismo pelo qual o paciente desenvolve lesão hepática não é conhecido –, é difícil identificar os pacientes sob risco. Em alguns casos, a impossibilidade de determinar o mecanismo ou mecanismos responsáveis pela lesão hepática deve-se à incapacidade de reproduzir a lesão em animais de laboratório. Outro desafio é o fato de que pode não ser possível prever a ocorrência de hepatotoxicidade com base nos estudos pré-clínicos, visto que os compostos que a exibem de maneira significativa em estudos de animais nas doses próximas à exposição terapêutica prevista em seres humanos geralmente são eliminados. Mais um fator que complica a prevenção da hepatotoxicidade é os estudos clínicos de um fármaco tipicamente incluírem milhares de pacientes, embora um risco de hepatotoxicidade induzida por fármaco na faixa de 1 em 10.000 a 1 em 100.000 pacientes seja preocupação suficiente para a retirada do fármaco. Em outras palavras, muitos estudos clínicos são demasiadamente pequenos, ou foram planejados com critérios de exclusão que não são mantidos após a comercialização do fármaco, a fim de detectar riscos inaceitáveis de hepatotoxicidade. Por exemplo, a retirada do mercado da troglitazona, agente sensibilizante da insulina, só ocorreu quando foi constatado que aproximadamente 1 em 10.000 pacientes tratados com esse fármaco falecia de insuficiência hepática aguda.
As atividades de certas enzimas no soro (alanina aminotransferase [ALT], aspartato aminotransferase [AST], fosfatase alcalina [ALP] e bilirrubina são usadas com frequência para monitorar o potencial de hepatotoxicidade nos pacientes. O melhor preditor de resultado para a hepatotoxicidade induzida por fármaco consiste na combinação de lesão hepatocelular (indicada por aumento na atividade sérica de ALT, AST e ALP) com diminuição da função hepática (indicada por níveis elevados de bilirrubina). A elevação dos níveis séricos de ALT em > 3 vezes o limite superior dos valores de referência, aliada à elevação do nível sérico de bilirrubina de > 2 vezes o limite superior do valor de referência, está associada a uma taxa de mortalidade de pelo menos 10%. Esse preditor tornou-se conhecido como regra de “Hy”, em homenagem ao hepatologista Hyman Zimmerman.
Toxicidade renal induzida por fármacos:
O rim constitui importante via de eliminação de muitos fármacos e seus metabólitos. A nefrotoxicidade pode manifestar-se como alterações da hemodinâmica renal, lesão e obstrução tubulares, nefropatia glomerular e nefrite intersticial. A insuficiência renal progressiva, caracterizada por aumentos crescentes dos níveis séricos de creatinina, pode resultar em perda de função de uma quantidade suficiente de néfrons. Exemplos de classes de fármacos passíveis de provocar insuficiência renal incluem certos antibióticos, AINE, agentes antineoplásicos, imunomoduladores e inibidores da enzima conversora de angiotensina (ECA). A seguir são descritos os mecanismos de nefrotoxicidade causados pelo antibiótico aminoglicosídio gentamicina e pelo agente antifúngico anfotericina B. A lesão renal constitui efeito adverso comum do tratamento com ambos os agentes.
A gentamicina provoca lesão renal em parte devido à inibição das hidrolases lisossômicas (esfingomielinases, fosfolipases) nos túbulos proximais do rim, resultando em acúmulo de estruturas lamelares eletrondensas contendo fosfolipídios não degradados nos lisossomos. Esse processo é denominado fosfolipidose renal. A ruptura dos lisossomos produz morte celular na forma de necrose tubular aguda. A lesão tubular causada pela gentamicina e por outros antibióticos aminoglicosídios é reversível com a interrupção do tratamento, contanto que a lesão inicial não seja muito grave. A anfotericina B provoca lesão das membranas celulares dos fungos ao interagir com o ergosterol e formar poros na membrana através dos quais o potássio sai, resultando em morte celular. A lesão renal induzida pela anfotericina parece ocorrer por um mecanismo semelhante, com ligação inicial do fármaco a esteróis nas membranas das células epiteliais dos túbulos renais. Como o mecanismo responsável pela eficácia do fármaco é compartilhado pelo mecanismo responsável por sua toxicidade, a margem entre a exposição necessária para uma atividade antifúngica e aquela que provoca lesão renal é pequena, ocasionando alta frequência de lesão renal em pacientes tratados com anfotericina B. Foram desenvolvidas formulações lipossomais do fármaco na tentativa de reduzir essa toxicidade e aumentar sua meia-vida plasmática. Quando a lesão inicial não é muito grave, a interrupção do tratamento com anfotericina frequentemente resulta em recuperação da função renal.
Os meios de contraste radiológicos são administrados por via intra-arterial ou intravenosa para definição radiográfica da vasculatura em determinados órgãos, como coração e cérebro. Esses agentes parecem causar lesão renal por toxicidade direta das células epiteliais dos túbulos renais e por constrição dos vasos retos, provocando diminuição do fluxo sanguíneo medular renal. A nefrotoxicidade dos meios de contraste radiológicos está relacionada com a dose, e os pacientes com redução preexistente do fluxo sanguíneo medular – devido, por exemplo, a insuficiência renal, depleção do volume intravascular, insuficiência cardíaca, diabetes melito ou uso de diuréticos ou AINE – correm maior risco.
Neurotoxicidade induzida por fármaco:
A neurotoxicidade induzida por fármacos está frequentemente mais associada a uso de agentes quimioterápicos para o câncer. Na maioria dos casos, manifesta-se nos nervos periféricos, porém o sistema nervoso central também pode ser afetado. A neuropatia periférica tem sido associada ao uso de alcaloides da vinca (p. ex., vincristina, vimblastina), taxanos (p. ex., paclitaxel) e compostos de platina (p. ex., cisplatina). A neuropatia provocada por alcaloides da vinca e por taxanos está diretamente relacionada com o mecanismo primário de ação desses fármacos, que consiste em ruptura dos microtúbulos. Nos nervos periféricos, acredita-se que essa ruptura resulte em alteração do tráfego axônico e neuropatia tanto sensorial quanto motora. Os compostos que contêm platina podem exercer efeitos tóxicos diretos sobre os nervos periféricos.
Toxicidade do músculo esquelético induzida por fármacos:
As classes de fármacos associadas a lesão do músculo esquelético incluem inibidores da HMG-CoA redutase (estatinas), corticosteroides (dexametasona, betametasona, prednisolona, hidrocortisona) e zidovudina (AZT ou ZDV). A miopatia induzida por estatinas parece estar relacionada com a inibição da geranil-geranilação de várias proteínas musculares. A lesão muscular induzida por corticosteroides é complexa, envolvendo alteração do metabolismo dos carboidratos, diminuição da síntese proteica e alterações da função mitocondrial que reduzem a capacidade oxidativa. Pacientes tratados com corticosteroides podem apresentar fraqueza, atrofia, mialgia e diminuição microscópica do tamanho das fibras musculares. Essa lesão é reversível, embora lentamente. A compreensão da patogenia da miopatia induzida pela zidovudina é complicada, em virtude da capacidade do HIV – infecção viral para a qual a zidovudina é administrada – de induzir miopatia na ausência de terapia farmacológica. Todavia, a melhora da função muscular observada com a interrupção da zidovudina e a demonstração independente de miopatia induzida por zidovudina em roedores sugerem que o próprio fármaco provoca miopatia, pelo menos em alguns pacientes. O mecanismo da miopatia associada à zidovudina não está bem elucidado, porém acredita-se que o acúmulo do fármaco no músculo esquelético, a ruptura das cristas mitocondriais e a diminuição da fosforilação oxidativa atuem nesse sentido.
Toxicidade cardiovascular induzida por fármacos:
Foram reconhecidos três mecanismos principais de toxicidade cardiovascular induzida por fármacos. Em primeiro lugar, conforme discutido anteriormente, muitos fármacos interagem com os canais de potássio cardíacos, causando prolongamento do QTc, repolarização tardia e arritmias cardíacas. Em segundo lugar, alguns fármacos são diretamente tóxicos para os miócitos cardíacos. Um agente antineoplásico antraciclínico, a doxorrubicina, liga-se de maneira intensa ao ferro; na presença de oxigênio, o ferro pode deslocar-se ciclicamente entre os estados de ferro (II) e ferro (III), acarretando produção de espécies reativas de oxigênio (ROS). Essas ROS promovem citotoxicidade e morte dos miócitos cardíacos, que apresentam baixa atividade dos sistemas enzimáticos antioxidantes. A cardiotoxicidade, resultando em insuficiência cardíaca e arritmias, frequentemente constitui a toxicidade que limita a dose em pacientes tratados com esse fármaco. Em terceiro lugar, conforme assinalado anteriormente, alguns fármacos são tóxicos para as valvas cardíacas. O análogo da anfetamina, fenfluramina, exerce seu efeito anorético desejado ao aumentar a liberação de serotonina e diminuir sua captação. A fenfluramina e seu metabólito, norfenfluramina, ligam-se também com alta afinidade aos receptores 5-HT2B. Essa ligação nas valvas cardíacas ativa as vias mitogênicas, induzindo proliferação dos miofibroblastos valvares que formam placas mixoides nas valvas atrioventriculares, produzindo insuficiência valvar e morte em alguns pacientes. A atividade da fenfluramina nos receptores 5-HT2B também pode aumentar a resistência vascular e remodelar o sistema arterial pulmonar, ocasionando desenvolvimento de hipertensão pulmonar. Devido à gravidade potencial dessas toxicidades cardiovasculares, existe um esforço combinado para evitar a seleção de compostos para desenvolvimento de fármacos capazes de provocar prolongamento significativo do intervalo QTc ou afinidade de ligação pelos receptores.
Toxicidade pulmonar induzida por fármacos:
Os efeitos adversos nos pulmões variam desde exacerbações agudas e reversíveis dos sintomas asmáticos até lesão crônica caracterizada por remodelagem e/ou fibrose. A obstrução reversível das vias respiratórias pode estar associada à terapia com agonistas β, enquanto se observa a ocorrência de lesão crônica em alguns pacientes que recebem o agente quimioterápico bleomicina ou o fármaco antiarrítmico amiodarona. A resposta à lesão após dano celular é determinada, em grande parte, pela capacidade de regeneração do órgão-alvo. Agressões repetidas ao pulmão, particularmente às células epiteliais que revestem as vias respiratórias de condução e os alvéolos, podem ser seguidas de regeneração. Ciclos repetidos de lesão epitelial podem induzir depósito excessivo de colágeno e proteínas da matriz extracelular em septos e espaços alveolares, causando fibrose. A fibrose pulmonar manifesta-se por perda de função. A bleomicina e a amiodarona estão contraindicadas para pacientes com doença do parênquima pulmonar, visto que ambas podem causar fibrose pulmonar.
Carcinogênese devida à terapia farmacológica:
Os fármacos (e outros agentes) que podem causar câncer são denominados carcinógenos. De modo mais geral, um carcinógeno é uma agressão química, física ou biológica que provoca tipos específicos de lesão do DNA (esses agentes são denominados iniciadores) ou facilita a proliferação de células com mutações pré-cancerosas (esses agentes são conhecidos como promotores). Os iniciadores atuam por meio de lesão do DNA, interferindo em sua replicação ou seus mecanismos de reparo. São, em sua maioria, espécies reativas, que modificam de modo covalente a estrutura do DNA, impedindo sua replicação acurada e, se não houver reparo ou se este for incorreto, resulta em uma ou mais mutações. Se a mutação ou mutações afetarem um ou mais genes que controlam a regulação do ciclo celular, poderá haver transformação neoplásica. A carcinogênese é um processo complexo, que envolve múltiplas alterações genéticas e epigenéticas e que habitualmente se estende por vários anos ou décadas.
Na maioria das áreas terapêuticas, evita-se o uso de compostos capazes de provocar lesão direta do DNA. Todavia, a lesão do DNA e/ou a interferência em seu reparo constituem o efeito terapêutico desejado de numerosos agentes usados no tratamento das neoplasias. A lesão das células sanguíneas progenitoras normais constitui importante efeito adverso no alvo de agentes alquilantes citotóxicos utilizados na quimioterapia do câncer (clorambucila, ciclofosfamida, melfalana, mostardas nitrogenadas e nitrosoureias). Esses fármacos podem causar mielodisplasia e/ou leucemia mieloide aguda (LMA). Com efeito, 10 a 20% dos casos de LMA nos EUA surgem secundariamente ao tratamento de outros cânceres com esses agentes antineoplásicos. O tamoxifeno, modulador do receptor de estrogênio não genotóxico, constitui tratamento efetivo para pacientes com câncer de mama sensíveis ao estrogênio. Todavia, esse fármaco também aumenta o risco de alguns tumores. Embora seja um antagonista dos receptores de estrogênio nas mamas, atua como agonista parcial em outros tecidos que expressam o receptor de estrogênio, em especial o útero. Logo, um efeito adverso do tratamento de câncer de mama com tamoxifeno pode consistir no desenvolvimento de câncer endometrial. Os novos moduladores dos receptores de estrogênio, como o raloxifeno, não estimulam os receptores de estrogênio uterinos, portanto podem ser usados no tratamento ou na prevenção do câncer de mama com menor risco de câncer endometrial.
As bulas de produtos descrevem a avaliação pré-clínica de cada fármaco na seção intitulada “Carcinogênese, Mutagênese, Comprometimento da Fertilidade”. Nessa seção, não é raro encontrar descrições de estudos realizados em roedores que sugerem o potencial carcinogênico dos fármacos. Como os fármacos desenvolvidos tipicamente não são mutágenos (salvo as exceções assinaladas anteriormente), os tumores relacionados com tratamento, observados nesses estudos longitudinais de roedores submetidos a altas doses do fármaco, em geral são atribuídos a mecanismos não genotóxicos (epigenéticos). Para avaliar se os achados em roedores representam risco para a população-alvo de pacientes, é importante compreender o mecanismo pelo qual esses tumores ocorrem. Por exemplo, o omeprazol, inibidor da bomba de prótons, provoca tumores das células enterocromafínicas-símiles (ECL) gástricas em roedores. O desenvolvimento desses tumores resulta de um aumento persistente e dose-relacionado da gastrina, que é secundário ao efeito desejado do composto (diminuição da secreção de ácido). Entretanto, a exposição necessária para a elevação prolongada da gastrina e a formação de tumores em roedores é muito maior que a exposição necessária para a eficácia do fármaco nos pacientes. Além disso, as elevações da gastrina observadas em pacientes são de baixa magnitude e não persistentes. Sendo assim, os achados carcinogênicos em estudos de roedores não são considerados como sinal de risco para o desenvolvimento de tumores em pacientes.
Teratogênese devida à terapia farmacológica:
Os fármacos administrados a gestantes podem afetar adversamente o feto. A teratogênese refere-se à indução de defeitos estruturais no feto, e um teratógeno é uma substância capaz de induzir esses defeitos. A exposição do feto a qualquer substância é determinada por absorção, distribuição, metabolismo e excreção maternas do fármaco e pela capacidade de o teratógeno ativo atravessar a placenta. Essas questões são discutidas de modo mais detalhado no Boxe 5.1. Fármacos que talvez exerçam poucos efeitos adversos sobre a mãe podem causar dano substancial no feto. Como o desenvolvimento do feto é, em termos cronológicos, precisamente programado, o efeito teratogênico de qualquer substância depende da fase de desenvolvimento em que ocorre a exposição. Nos seres humanos, a organogênese geralmente ocorre entre a terceira e oitava semanas de gestação, e é durante esse período que os teratógenos exercem seus efeitos mais profundos. Antes da terceira semana, os compostos tóxicos produzem, em sua maioria, morte do embrião e aborto espontâneo, ao passo que, depois da organogênese, os compostos teratogênicos podem afetar o crescimento e a maturação funcional dos órgãos, porém não afetam o plano básico de desenvolvimento. Por exemplo, o ácido retinoico (vitamina A) apresenta significativa toxicidade teratogênica no alvo. Ele ativa os receptores retinoides nucleares (RAR) e os receptores X retinoides (RXR), que regulam diversos eventos essenciais da transcrição durante o desenvolvimento. Tendo em vista a gravidade dos defeitos congênitos que podem ocorrer, as mulheres em uso de agonistas dos RAR/RXR, como a isotretinoína, para o tratamento da acne devem assinar formulários de consentimento esclarecedores exigidos pela FDA para demonstrar que estão cientes do risco de defeitos congênitos graves relacionados com o uso do fármaco.
Outro exemplo de efeito teratogênico no alvo é a exposição in utero do feto a inibidores da ECA. Embora estes não estivessem anteriormente contraindicados no primeiro trimestre de gravidez, dados recentes indicam que a exposição do feto durante esse período aumenta de maneira significativa os riscos de malformações dos sistemas cardiovascular e nervoso central. Os inibidores da ECA podem causar um conjunto de afecções, incluindo oligoidrâmnio, retardo do crescimento intrauterino, displasia renal, anúria e insuficiência renal, refletindo a importância da via da angiotensina sobre o desenvolvimento e a função renais.
Princípios para tratamento dos pacientes com toxicidade induzida por fármacos:
O tratamento da toxicidade induzida por fármacos pode incluir: redução ou eliminação da exposição ao fármaco; administração de tratamentos específicos baseados no antagonismo ao mecanismo de ação do fármaco ou na alteração de seu metabolismo; e/ou medidas de suporte. A redução da exposição a um agente terapêutico em um paciente que apresenta efeitos adversos pode parecer natural, porém nem sempre representa a escolha correta. O aparecimento de um efeito adverso durante a terapia não significa necessariamente que seja causado pelo fármaco, apesar da relação temporal entre o início do tratamento e o aparecimento do efeito adverso. Embora esse efeito tenha ocorrido mais provavelmente em consequência do uso do fármaco, os riscos de sua interrupção precisam ser avaliados em relação aos benefícios de sua continuação. Obviamente, a interrupção da terapia é uma escolha correta quando os efeitos adversos foram anteriormente associados ao fármaco e são potencialmente fatais, como a anafilaxia causada por um antibiótico betalactâmico. É desnecessário dizer, entretanto, que, para esses pacientes, qualquer tratamento futuro com essa classe de antibióticos estaria contraindicado. Os efeitos adversos irreversíveis e/ou cuja gravidade tende a aumentar com o tratamento continuado também podem motivar a decisão apropriada de interromper o tratamento. Entretanto, muitos efeitos adversos são considerados toleráveis e reversíveis. Dependendo da gravidade da doença tratada, é possível que o benefício global para o paciente seja maior com o tratamento farmacológico do que sem ele. Um exemplo dessa situação é a leucopenia que ocorre com frequência em pacientes submetidos à quimioterapia com agentes citotóxicos. Por conseguinte, a decisão de interromper ou reduzir o tratamento pode ser complexa e, com frequência, exige avaliação de muitos fatores que afetam a saúde imediata e a longo prazo do paciente.
Os tratamentos destinados a neutralizar os efeitos adversos produzidos por determinado fármaco baseiam-se, com frequência, no antagonismo de sua atividade farmacodinâmica ou na interferência dos efeitos relacionados com a sua farmacocinética. O antagonismo da atividade farmacológica de um fármaco constitui abordagem útil para as superdosagens de opioides, benzodiazepínicos e inibidores da acetilcolinesterase (AChE). A interferência nos efeitos tóxicos dos metabólitos de um fármaco constitui abordagem útil ao tratamento da toxicidade do paracetamol. Esses exemplos serão brevemente discutidos a seguir. Em termos conceituais, o tratamento mais simples da superdosagem de um fármaco consiste na administração de um antagonista capaz de bloquear a ação do fármaco que, direta ou indiretamente, resulta em ativação suprafisiológica de um receptor. Por exemplo, uma superdosagem de opioides pode ser tratada com naloxona, antagonista farmacológico do receptor de opioides. Ligando-se competitivamente aos receptores opioides, a naloxona impede ou reverte os efeitos tóxicos dos opioides naturais ou sintéticos, os quais incluem depressão respiratória, sedação e hipotensão. Esse fármaco começa a agir com rapidez e é superpotente; com efeito, se não for observada nenhuma melhora clínica dentro de 10 min após a administração de doses dele até 10 mg, devem-se considerar um diagnóstico diferente ou múltiplas entidades tóxicas. A naloxona apresenta meia-vida relativamente curta, de modo que precisa ser administrada a cada uma a quatro horas para proporcionar antagonismo adequado dos receptores enquanto o opioide está sendo depurado.
O flumazenil, antagonista farmacológico do receptor de GABAA (benzodiazepínico), é utilizado no tratamento da superdosagem de benzodiazepínicos. Ele atua por inibição competitiva nos receptores de benzodiazepínicos no sistema nervoso central, revertendo de modo completo ou parcial os efeitos sedativos desses fármacos. À semelhança da naloxona, o flumazenil começa a agir com rapidez e é superpotente; seus efeitos devem ser observados dentro de cinco minutos após a administração de uma dose de no máximo 3 mg. Também tem meia-vida curta (de aproximadamente uma hora) e precisa ser administrado a intervalos frequentes para proporcionar antagonismo adequado dos receptores enquanto o benzodiazepínico está sendo depurado.
Pode-se utilizar também o antagonismo farmacológico quando o agente tóxico não é um agonista direto, porém aumenta indiretamente a concentração do ligante natural de um receptor. Os inibidores da AChE produzem concentração suprafisiológica de acetilcolina na fenda sináptica e toxíndrome característica de excesso colinérgico – bradicardia, miose, hipersalivação, sudorese, diarreia, vômitos, broncoconstrição, fraqueza, paralisia respiratória e convulsões. Embora seja algumas vezes possível restaurar a atividade da AChE, o tratamento de sua inibição depende, em geral, da administração de um agente anticolinérgico, como a atropina. Esta, que antagoniza o receptor muscarínico de acetilcolina, restaura o equilíbrio colinérgico e impede a broncoconstrição, causa mais comum de morte em pacientes expostos a inibidores da AChE.
Conforme assinalado anteriormente, uma das consequências da superdosagem de paracetamol consiste na depleção da glutationa intracelular pelo metabólito do fármaco, a N-acetil-pbenzoquinoneimina (NAPQI). É possível repor as reservas de glutationa pela administração de N- ▶ acetilcisteína (NAC), precursor metabólico da glutationa (Figura 5.4). Além da terapia de suporte (lavagem gástrica e/ou carvão ativado), a NAC é administrada por via oral ou intravenosa dentro de 8 a 10 h após a ingestão de uma dose potencialmente hepatotóxica de paracetamol para evitar ou diminuir a lesão hepática.
Por fim, pode-se fornecer uma terapia de suporte em caso de toxicidade induzida por fármacos. Exemplo disso é a administração de líquidos intravenosos a pacientes com lesão renal, a fim de manter um fluxo sanguíneo renal adequado. Em caso de lesão renal grave, a hemodiálise pode tornarse necessária até a recuperação da função renal. Outro exemplo é o tratamento da supressão da medula óssea que resulta da administração de agentes citotóxicos na quimioterapia do câncer. O filgrastim, fator de estimulação de colônias de granulócitos (G-CSF) humano recombinante, pode ser utilizado para estimular a produção de leucócitos e fornecer terapia de suporte até a recuperação da produção endógena de leucócitos da medula óssea com o término da terapia citotóxica.
Conclusão e perspectivas:
Este capítulo apresentou uma abordagem baseada em mecanismos para compreender a toxicidade farmacológica e forneceu exemplos para ilustrar esses princípios nos sistemas orgânicos de maior importância. As metas no desenvolvimento de fármacos continuam sendo a descoberta de compostos ao mesmo tempo efetivos e altamente seletivos, portanto com menos probabilidade de causar efeitos graves ou indesejáveis fora do alvo terapêutico. Os desafios do futuro residem particularmente na compreensão da base da variabilidade das respostas terapêuticas e tóxicas às substâncias. Em uma tentativa de prever quais populações de pacientes serão mais suscetíveis a determinada reação farmacológica adversa, uma das abordagens em avaliação consiste em identificar correlações entre polimorfismos de nucleotídios simples (SNP) individuais e possíveis reações adversas, comparando os SNP dos pacientes que apresentam reações adversas com os indivíduos que não as apresentam. A identificação de pacientes com variantes genéticas do alvo molecular (e alvos estreitamente relacionados) de um fármaco também pode fornecer informações úteis sobre os indivíduos com mais tendência a apresentar efeitos adversos.
Prever a eficácia e a segurança de um fármaco em determinado paciente continua sendo um desafio para o médico. A decisão quanto ao uso de terapia farmacológica exige o conhecimento dos benefícios e riscos potenciais dessa terapia. Além disso, os médicos têm a responsabilidade de comunicar esses riscos e benefícios ao paciente, de modo que possa ser considerada toda a gama de opções terapêuticas. Um dos maiores desafios para o médico é saber onde encontrar essas informações. As fontes incluem a literatura científica, o rótulo do produto, as comunicações diretas entre paciente e médico e a revisão dos dados pré-clínicos e clínicos preparados pela FDA durante sua revisão de uma New Drug Application. As principais informações sobre toxicidade, tanto pré-clínicas como clínicas, são fornecidas na bula do produto, cujas revisões a respeito podem ser efetuadas à medida que eventos adversos graves são atribuídos ao fármaco durante a vigilância pós-comercialização, e cabe ao médico consultar a versão mais atualizada da bula do produto. Advertências sobre consequências graves também podem ser transmitidas por meio de comunicações diretas aos médicos, e o website da FDA pode ser consultado para medidas regulamentares relacionadas com a segurança de um fármaco. O website da European Medicines Agency (EMEA) contém informações sobre medidas regulamentares para medicamentos comercializados na Europa. A Tabela 5.2 fornece uma lista de algumas fontes online que podem ser consultadas para informações mais detalhadas sobre toxicidade farmacológica. Boas fontes de informações detalhadas sobre toxicidade pré-clínica e eventos adversos clínicos são documentos preparados por farmacologista da FDA (pré-clínico) e revisor médico (clínico) como parte da revisão da NDA.