Metabolismo dos Fármacos
NOÇÕES BÁSICAS EM FARMACOLOGIA
1 Fármacos:
Nossos tecidos são expostos todos os dias a xenobióticos – substâncias estranhas não encontradas naturalmente no corpo. Os fármacos são, em sua maioria, xenobióticos utilizados para modular funções corporais com fins terapêuticos. Eles e outras substâncias químicas ambientais que penetram no organismo são modificados por uma enorme variedade de enzimas. As transformações bioquímicas efetuadas por essas enzimas podem alterar o composto, tornando-o benéfico, prejudicial ou simplesmente ineficaz. Os processos pelos quais as reações bioquímicas alteram os fármacos no corpo são designados, em seu conjunto, como metabolismo ou biotransformação dos fármacos. O capítulo anterior introduziu a importância da depuração renal na farmacocinética dos fármacos. Embora as reações bioquímicas que modificam os fármacos, convertendo-os em formas passíveis de excreção renal, constituam parte essencial do metabolismo dos fármacos, tal metabolismo abrange mais do que essa função. A biotransformação pode alterá-los de quatro maneiras:
- Um fármaco ativo pode ser convertido em um fármaco inativo
- Um fármaco ativo pode ser convertido em um metabólito ativo ou tóxico
- Um profármaco inativo pode ser convertido em um fármaco ativo
- Um fármaco não passível de excreção pode ser convertido em um metabólito passível de excreção (p. ex., aumentando a depuração renal ou biliar).
Este capítulo descreve os principais processos de metabolismo dos fármacos. Após a apresentação do caso, o capítulo fornece uma visão geral dos locais de metabolismo dos fármacos, enfocando principalmente o fígado. A seguir, são discutidos os dois tipos principais de biotransformação, denominados, com frequência, reações de fase I e de fase II, embora a terminologia seja imprecisa e implique, de modo incorreto, uma ordem cronológica das reações. (Além disso, utiliza-se algumas vezes o termo fase III para descrever o processo de transporte dos fármacos, produzindo ainda mais confusão.) Neste capítulo, são empregadas as expressões oxidação/redução e conjugação/hidrólise para descrever esses processos de modo mais acurado. O capítulo termina com uma discussão dos fatores que podem resultar em diferenças no metabolismo dos fármacos entre indivíduos distintos.
Locais de metabolismo dos fármacos:
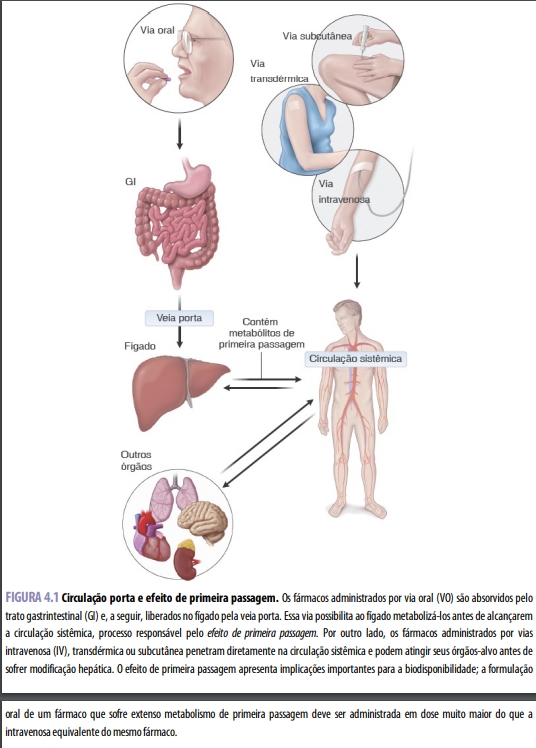
O fígado é o principal órgão de metabolismo dos fármacos. Esse fato evidencia-se de maneira proeminente no fenômeno conhecido como efeito de primeira passagem. Com frequência, os fármacos administrados por via oral são absorvidos no trato gastrintestinal (GI) e transportados diretamente até o fígado por meio da circulação porta. Assim, o fígado tem a oportunidade de metabolizá-los antes de alcançarem a circulação sistêmica, portanto antes de atingirem seus órgãos-alvo. É preciso considerar o efeito de primeira passagem quando se planejam 1. 2. 3. esquemas posológicos, visto que, se o metabolismo hepático for extenso, a quantidade de fármaco que alcançará o tecido-alvo será muito menor do que a quantidade (dose) administrada por via oral. Certos fármacos são inativados de modo tão eficiente em sua primeira passagem pelo fígado que não podem ser administrados por via oral, devendo-se utilizar a via parenteral. Um deles é o agente antiarrítmico lidocaína, cuja biodisponibilidade é de apenas 3% quando administrado por via oral.
Embora o fígado seja quantitativamente o órgão mais importante no metabolismo dos fármacos, todos os tecidos do corpo são capazes de metabolizá-los, em certo grau. Os locais ativos em particular incluem pele, pulmões, trato gastrintestinal e rins. O trato gastrintestinal merece menção especial, visto que, à semelhança do fígado, pode contribuir para o efeito de primeira passagem mediante o metabolismo dos fármacos administrados por via oral antes que alcancem a circulação sistêmica.
Vias de metabolismo dos fármacos:
Fármacos e outros xenobióticos sofrem biotransformação antes de serem excretados. Muitos produtos farmacêuticos são lipofílicos, o que possibilita ao fármaco atravessar as membranas celulares, como as encontradas na mucosa intestinal ou no tecido-alvo. Infelizmente, a mesma propriedade química que aumenta a biodisponibilidade dos fármacos também pode dificultar sua excreção renal, visto que a depuração pelo rim exige que esses fármacos tornem-se mais hidrofílicos, de modo que possam ser dissolvidos na urina aquosa. Por conseguinte, as reações de biotransformação frequentemente aumentam a hidrofilia dos compostos para torná-los mais passíveis de excreção renal. As reações de biotransformação são classicamente divididas em dois tipos principais: oxidação/redução (fase I) e conjugação/hidrólise (fase II). Em geral, as reações de oxidação transformam o fármaco em metabólitos mais hidrofílicos pela adição de ou exposição a grupos funcionais polares, como grupos hidroxila (-OH) ou amina (-NH2) (Tabela 4.1). Com frequência, esses metabólitos são farmacologicamente inativos e podem ser secretados sem qualquer modificação adicional. Entretanto, alguns produtos de reações de oxidação e redução necessitam de modificações adicionais antes de serem excretados. As reações de conjugação (fase II) modificam os compostos por meio da ligação de grupos hidrofílicos, como o ácido glicurônico, criando conjugados mais polares (Tabela 4.2). É importante assinalar que essas reações ocorrem independentemente das reações de oxidação/redução, e que as enzimas envolvidas nas reações de oxidação/redução e de conjugação/hidrólise frequentemente competem pelos substratos.
Reações de oxidação/redução:
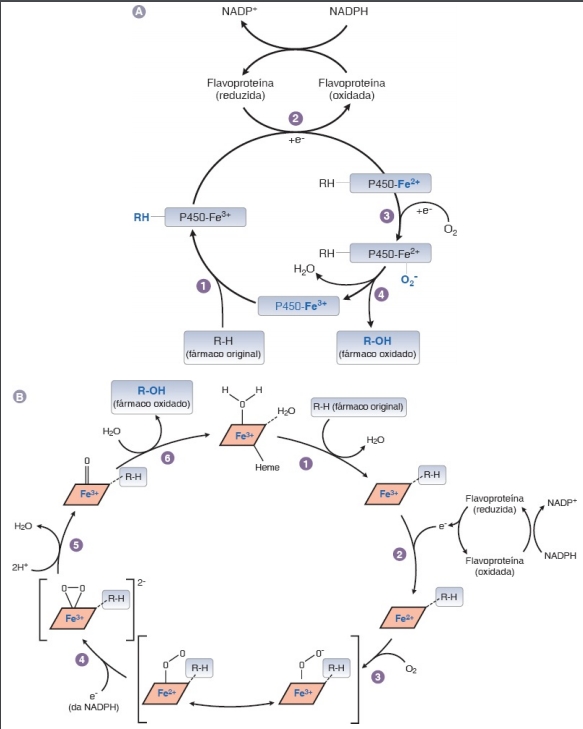
As reações de oxidação envolvem enzimas associadas a membranas, expressas no retículo endoplasmático (RE) dos hepatócitos e, em menor grau, de células de outros tecidos. As enzimas que catalisam essas reações de fase I são tipicamente oxidases. São, em sua maioria, hemeproteínas mono-oxigenases da classe do citocromo P450. As enzimas do citocromo P450 (algumas vezes abreviadas CYP) são também conhecidas como oxidases microssomais de função mista e são envolvidas no metabolismo de aproximadamente 75% de todos os fármacos utilizados hoje. (O termo P450 refere-se à característica de pico de absorção em 450 nm dessas hemeproteínas quando se ligam ao monóxido de carbono.) O resultado de uma reação de oxidação que depende do citocromo P450 é o seguinte:
A reação prossegue quando a substância liga-se ao citocromo P450 oxidado (Fe 3+ ) para formar um complexo, depois reduzido em duas etapas sequenciais de oxirredução. A nicotinamida adenina dinucleotídeo fosfato (NADPH) doa os elétrons nas duas etapas via uma flavoproteína redutase. Na primeira etapa, o elétron doado reduz o complexo citocromo P450-fármaco. Na segunda, o elétron reduz o oxigênio molecular para formar um complexo oxigênio-citocromo P450- fármaco ativado. Por fim, à medida que o complexo torna-se mais ativo graças ao rearranjo, o átomo de oxigênio reativo é transferido para o fármaco, resultando na formação de um produto oxidado de fármaco e na reciclagem do citocromo P450 oxidado no processo
A maioria das oxidases hepáticas do citocromo P450 apresenta ampla especificidade de substrato. Isso se deve, em parte, ao oxigênio ativado do complexo, que é um potente agente oxidante e reage com vários tipos de substrato. As enzimas do citocromo P450 são, às vezes, designadas “P450” seguido do número da família de enzimas P450, da letra maiúscula da subfamília e de um número adicional para identificar a enzima específica. Muitas enzimas P450 exibem especificidades parcialmente superpostas que, em seu conjunto, possibilitam ao fígado reconhecer e metabolizar uma gama de xenobióticos. Em seu conjunto, as reações mediadas pelo P450 respondem por mais de 95% das biotransformações oxidativas. Outras vias também podem oxidar moléculas lipofílicas. Exemplo pertinente de uma via oxidativa não P450 é a via da álcool desidrogenase, que oxida os álcoois a seus derivados aldeídicos como parte do processo global de excreção. Essas enzimas constituem a base da toxicidade do metanol. Este é oxidado pela álcool desidrogenase a formaldeído, que causa considerável dano a alguns tecidos. O nervo óptico mostra-se particularmente sensível ao formaldeído, e a toxicidade do metanol pode causar cegueira. Outra enzima não P450 importante é a monoamina oxidase (MAO), responsável pela oxidação de compostos endógenos que contêm amina, como as catecolaminas e a tiramina, e de alguns xenobióticos, incluindo fármacos.
Reações de conjugação/hidrólise:
As reações de conjugação e hidrólise proporcionam um segundo conjunto de mecanismos destinados a modificar os compostos para sua excreção (Figura 4.3). Embora a hidrólise de fármacos que contêm éster e amida seja algumas vezes incluída entre as reações de fase I (na antiga terminologia), a bioquímica da hidrólise está mais estreitamente relacionada com a conjugação do que com a oxidação/redução. Os substratos dessas reações englobam tanto metabólitos de reações de oxidação (p. ex., epóxidos) quanto compostos que já contêm grupos químicos apropriados para conjugação, como hidroxila (-OH), amina (-NH2) ou carboxila (-COOH). Esses substratos são acoplados a metabólitos endógenos (p. ex., ácido glicurônico e seus derivados, ácido sulfúrico, ácido acético, aminoácidos e o tripeptídio glutationa) por enzimas de transferência, em reações que envolvem com frequência intermediários de alta energia. As enzimas de conjugação e de hidrólise localizam-se tanto no citosol quanto no retículo endoplasmático dos hepatócitos (e de outros tecidos). Na maioria dos casos, o processo de conjugação torna o fármaco mais polar. Praticamente todos os produtos conjugados são farmacologicamente inativos, com algumas exceções importantes (p. ex., glicuronídio de morfina).
Algumas reações de conjugação são clinicamente relevantes no caso dos recém-nascidos, que ainda não desenvolveram plenamente a capacidade de realizar esse conjunto de reações. A UDPglicuronil transferase (UDPGT) é responsável pela conjugação da bilirrubina no fígado, facilitando sua excreção. A deficiência de desenvolvimento dessa enzima no nascimento coloca os lactentes em risco de ter icterícia neonatal, que resulta da elevação dos níveis séricos de bilirrubina não conjugada. A icterícia neonatal representa um problema, visto que recém-nascidos não só apresentam atividade subdesenvolvida dessa enzima, como também uma barreira hematencefálica ainda não desenvolvida. A bilirrubina não conjugada, insolúvel em água e muito lipofílica, liga-se com facilidade ao cérebro desprotegido do recém-nascido e tem a capacidade de provocar lesão significativa do sistema nervoso central. Essa condição patológica é conhecida como encefalopatia por bilirrubina, ou kernicterus. A hiperbilirrubinemia neonatal (não conjugada) pode ser corrigida com fototerapia (comprimento de luz: 450 nm) que converte a bilirrubina circulante em isômero excretado mais rapidamente. Outro tratamento efetivo consiste na administração de doses pequenas do barbitúrico fenobarbital, que suprarregula de modo intenso a expressão da enzima UDPGT, reduzindo, assim, os níveis séricos de bilirrubina não conjugada. Isso ilustra o tema recorrente: a compreensão do metabolismo da substância pode ajudar a prever as interações medicamentosas adversas e as potencialmente vantajosas.
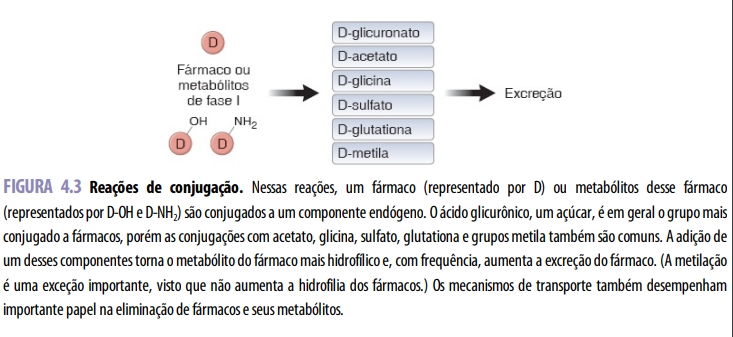
É importante assinalar que as reações de conjugação e de hidrólise não constituem, necessariamente, a última etapa de biotransformação. Como a conjugação desses componentes altamente polares ocorre no interior da célula, eles frequentemente precisam atravessar as membranas celulares por transporte ativo para serem excretados. (Pode ocorrer também transporte ativo do fármaco original.) Além disso, alguns produtos de conjugação podem sofrer metabolismo adicional.
2 Transporte de fármacos:
Embora muitos fármacos sejam lipofílicos o suficiente para atravessar de maneira passiva as membranas celulares, sabe-se hoje que muitos também precisam ser transportados ativamente para o interior das células. Esse fato produz consequências significativas para biodisponibilidade oral (transporte nos enterócitos ou excreção ativa no lúmen intestinal), metabolismo hepático (transporte nos hepatócitos para metabolismo enzimático e excreção na bile) e depuração renal (transporte nas células tubulares proximais e excreção no lúmen tubular). Tais processos são mediados por diversas moléculas importantes. A proteína de resistência a múltiplos fármacos 1 (MDR1, multidrug resistance protein 1) ou P-glicoproteína, membro da família ABC de transportadores de efluxo, transporta ativamente compostos de volta ao lúmen intestinal. Esse processo limita a biodisponibilidade oral de vários fármacos importantes, incluindo digoxina e inibidores da protease do HIV-1. Com frequência, o metabolismo dos fármacos na circulação porta (i. e., efeito de primeira passagem) exige o transporte de compostos nos hepatócitos por meio da família de proteínas do polipeptídio transportador de ânions orgânicos (OATP, organic anion transporting polypeptide) e transportador de cátions orgânicos (OCT, organic cation transporter). Esses transportadores são particularmente relevantes no metabolismo de vários inibidores redutase (estatinas) da 3-hidroxi-3- metilglutaril coenzima A (HMG-CoA), utilizados no tratamento da hipercolesterolemia. Por exemplo, o metabolismo do inibidor da HMG-CoA redutase, a pravastatina, depende do transportador OATP1B1, que transporta o fármaco nos hepatócitos. Acredita-se que a captação do fármaco nos hepatócitos por meio do OATP1B1 seja a etapa que limita a velocidade no processo de depuração da pravastatina. A captação desta em sua primeira passagem pelo fígado também representa vantagem potencial, visto que mantém o fármaco fora da circulação sistêmica, a partir da qual poderia ser captado pelas células musculares, causando efeitos tóxicos, como rabdomiólise. A família de transportadores de ânions orgânicos (OAT) é responsável pela secreção renal de muitas substâncias aniônicas clinicamente importantes, como os antibióticos betalactâmicos, os anti-inflamatórios não esteroides (AINE) e os antivirais análogos de nucleosídios.
Indução e inibição:
O uso do fenobarbital para evitar icterícia neonatal demonstra que o metabolismo dos fármacos pode ser influenciado pelos níveis de expressão das enzimas metabolizadoras. Embora algumas enzimas P450 sejam constitutivamente ativas, outras podem ser induzidas ou inibidas por diferentes compostos. A indução ou a inibição podem ser incidentais (efeito adverso de um fármaco) ou deliberadas (efeito desejado da terapia). O mecanismo primário da indução da enzima P450 é o aumento da expressão da enzima principalmente por transcrição aumentada, embora possa haver menor participação de aumento da translação e redução da degradação. A indução das enzimas P450 por uma gama de fármacos reflete a biologia dos receptores xenobióticos que atuam como sistema de vigilância do corpo para metabolizar compostos potencialmente tóxicos. Fármacos, poluentes ambientais, compostos químicos industriais e até mesmo alimentos podem penetrar nos hepatócitos e ligar-se a vários receptores xenobióticos diferentes, como receptor de pregnano X (PXR), receptor de androstano constitutivamente ativo (CAR) e receptor de aril-hidrocarboneto (AhR) (Figura 4.4). Essas moléculas são receptores nucleares de hormônios. Quando um composto xenobiótico liga-se ao receptor e ativa-o, o complexo é translocado para o núcleo e, aí, liga-se aos promotores de várias enzimas de biotransformação, acarretando aumento da expressão das enzimas P450. Por um mecanismo semelhante, a ativação do receptor nuclear de hormônio também pode aumentar a expressão dos transportadores de fármacos que ajudam na depuração dos compostos do corpo, como MDR1 e OATP1
A indução das enzimas P450 tem múltiplas consequências. Em primeiro lugar, o fármaco pode aumentar seu próprio metabolismo. Por exemplo, a carbamazepina, um antiepiléptico, não apenas induz a 3A4 do P450, como também é metabolizada por essa enzima. Logo, a carbamazepina acelera seu próprio metabolismo por meio da indução de 3A4 do P450. Em segundo lugar, um fármaco pode aumentar o metabolismo de outro fármaco coadministrado. Por exemplo, a 3A4 do P450 é responsável pelo metabolismo de mais de 50% de todos os fármacos clinicamente prescritos. Se um desse tipo for coadministrado com a carbamazepina, seu metabolismo também será aumentado. Essa situação pode ser problemática, visto que o aumento da atividade da 3A4 do P450 pode reduzir as concentrações do fármaco a níveis inferiores aos terapêuticos, se administradas doses convencionais desses fármacos. No caso da Srta. B, a administração de rifampicina com a terapia anti-HIV pode ser prejudicial, visto que essa substância induz a 3A4 do P450, aumentando, assim, o metabolismo de inibidores da protease, como o saquinavir. Em terceiro lugar, a indução das enzimas do P450 ou de algumas das outras enzimas de biotransformação pode resultar na produção de níveis tóxicos dos metabólitos reativos dos fármacos, produzindo lesão tecidual ou outros efeitos adversos.
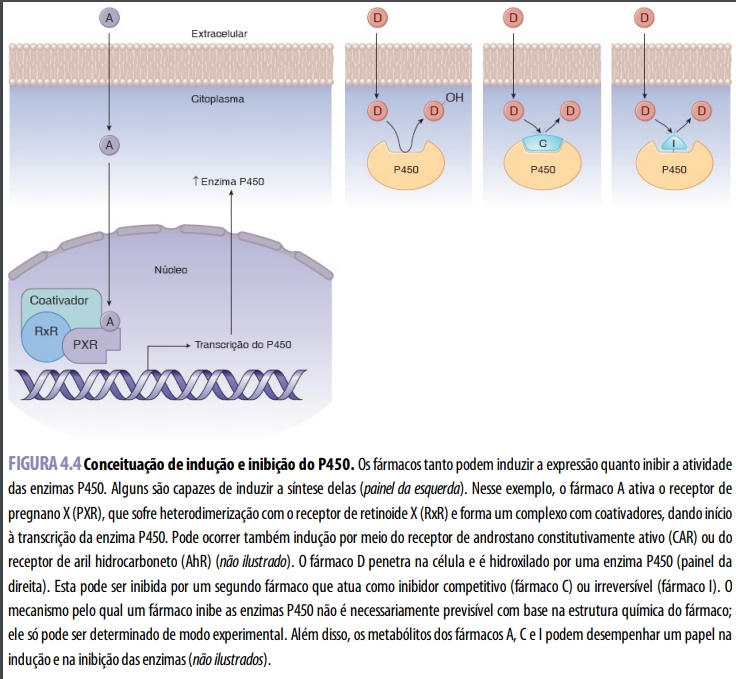
Assim como certos compostos podem induzir as enzimas P450, outros são capazes de inibi-las. Uma importante consequência da inibição de enzimas é a redução do metabolismo dos fármacos metabolizados pela enzima inibida. Essa inibição pode fazer com que os níveis do fármaco alcancem concentrações tóxicas e também pode prolongar a presença do fármaco ativo no corpo. A inibição enzimática pode ser obtida de diversas maneiras (Figura 4.4). Por exemplo, o cetoconazol, agente antifúngico amplamente utilizado, apresenta um nitrogênio que se liga ao ferro heme no sítio ativo das enzimas P450; essa ligação impede o metabolismo de fármacos coadministrados por inibição competitiva. Um exemplo de inibição irreversível é o secobarbital, barbitúrico que alquila e inativa permanentemente o complexo P450. Em certas ocasiões, a inibição das enzimas P450 pode produzir vantagem terapêutica. Por exemplo, o ritonavir (inibidor da protease) é extremamente eficaz contra o HIV, mas provoca efeitos adversos gastrintestinais significativos que limitam seu uso terapêutico como tratamento crônico. Todavia, como é um potente inibidor de P450 3A4, pode ser usado clinicamente em doses inferiores ao limiar dos efeitos adversos gastrintestinais (mas suficientemente elevadas para inibir P450 3A4. Essa inibição “reforça” as concentrações efetivas de outros inibidores da protease (IP) metabolizados por essa isoforma de P450. Por exemplo, o lopinavir não consegue atingir níveis terapêuticos quando utilizado como agente único, em virtude de seu extenso metabolismo de primeira passagem; entretanto, sua coadministração com ritonavir possibilita-lhe alcançar concentrações terapêuticas. Os transportadores de fármacos também podem ser induzidos ou inibidos por outros fármacos. Assim, por exemplo, os antibióticos macrolídeos são capazes de inibir o MDR1, e essa inibição pode levar a níveis séricos elevados de fármacos, como a digoxina, excretados pelo MDR1. Este também tem sua transcrição regulada por PXR. Como consequência, os fármacos que induzem a suprarregulação das enzimas P450 pela via do PXR (p. ex., P450 3A4) aumentam concomitantemente a transcrição do transportador MDR1.
Metabólitos ativos e tóxicos:
O conhecimento das vias pelas quais os agentes terapêuticos são metabolizados pode afetar a escolha do fármaco prescrito para determinada situação clínica. Isso se aplica tanto à situação em que o metabólito é ativo, quando o agente administrado pode atuar como profármaco, quanto à situação em que o fármaco apresenta metabólitos tóxicos. Os profármacos são compostos inativos metabolizados pelo corpo em suas formas terapêuticas ativas. Exemplo de profármaco é o tamoxifeno, modulador seletivo dos receptores de estrogênio; ele apresenta pouca atividade até sofrer hidroxilação, produzindo 4-hidroxitamoxifeno, metabólito 30 a 100 vezes mais ativo do que o composto original. Outro exemplo é o losartana, antagonista dos receptores de angiotensina II; a potência desse fármaco aumenta 10 vezes com a oxidação de seu grupo álcool para ácido carboxílico pela P450 2C9. A estratégia de ativação seletiva de profármacos pode ser utilizada com benefício terapêutico na quimioterapia do câncer. Um exemplo é o emprego de mitomicina C, composto de ocorrência natural, ativado a um poderoso agente alquilante do DNA após ser reduzido por várias enzimas, incluindo uma redutase do citocromo P450. A mitomicina C mata de maneira seletiva as células cancerosas hipóxicas na parte central de tumores sólidos, visto que: (1) essas células apresentam níveis elevados da redutase do citocromo P450, que ativa a mitomicina C; e (2) a reoxidação do fármaco é inibida em condições hipóxicas.
Fatores individuais que afetam o metabolismo dos fármacos:
Devido a diversos fatores, as velocidades das reações de biotransformação podem variar acentuadamente de uma pessoa para outra. Entre esses fatores, os mais importantes são discutidos a seguir.
Farmacogenômica:
Os efeitos da variabilidade genética sobre o metabolismo dos fármacos constituem importante parte da nova ciência da farmacogenômica (ver Capítulo 6). Determinadas populações exibem polimorfismos ou mutações em uma ou mais enzimas do metabolismo de fármacos, modificando a velocidade de algumas dessas reações e eliminando outras por completo. Essas diferenças farmacogenéticas precisam ser levadas em conta na tomada de decisões terapêuticas e na prescrição de doses dos medicamentos. Pesquisas atuais utilizam novas tecnologias (p. ex., análise SNP, microchips gênicos) para compreender como diferenças genéticas nas enzimas do metabolismo de fármacos influenciam a resposta dos pacientes a várias substâncias. Essas abordagens já são muito empregadas na elaboração de fármacos e estão começando a ser aplicadas na prática clínica. Por exemplo, a maioria das companhias farmacêuticas evita a criação de uma substância que seja metabolizada por uma enzima extremamente polimórfica porque esses polimorfismos resultam em grande variabilidade de resposta de um indivíduo para outro.
Outro importante exemplo clínico de variabilidade farmacogenética envolve a enzima plasmática colinesterase. Um em cada 2.000 caucasianos é portador de uma alteração genética nessa enzima, que metaboliza um relaxante muscular, a succinilcolina (entre outras funções). Essa forma alterada da enzima apresenta redução de afinidade pela succinilcolina de cerca de 1.000 vezes, ocasionando eliminação mais lenta e em circulação prolongada do fármaco ativo. Caso seja alcançada concentração plasmática de succinilcolina suficientemente alta, podem ocorrer paralisia respiratória e morte, a não ser que o paciente receba suporte com respiração artificial até que ocorra a depuração do fármaco. Situação semelhante é provável com isoniazida, um dos fármacos considerados no tratamento da tuberculose da Srta. B. A variabilidade genética, na forma de traço autossômico recessivo disseminado, que resulta em diminuição da síntese de uma enzima, causa retardo do metabolismo desse fármaco em certos subgrupos da população dos EUA. A enzima em questão é a Nacetiltransferase, que inativa a isoniazida por uma reação de acetilação (conjugação). O fenótipo de “acetilador lento” é expresso em 45% dos indivíduos brancos e negros nos EUA e por alguns europeus que vivem em altas latitudes norte. O fenótipo de “acetilador rápido” é encontrado em mais de 90% dos asiáticos e nos inuítes dos EUA. Os níveis sanguíneos de isoniazida estão de quatro a seis vezes mais elevados nos acetiladores lentos do que nos rápidos. Além disso, já que o fármaco livre atua como inibidor das enzimas P450, os acetiladores lentos estão mais sujeitos a interações medicamentosas adversas. Caso a Srta. B expresse o fenótipo de acetilador lento, e sua dose de isoniazida não seja diminuída com base nesse fato, a adição dessa substância a seu esquema posológico poderá potencialmente produzir efeito tóxico.
Um terceiro exemplo é o clopidogrel, antiplaquetário que promove a passagem do sangue nos vasos sanguíneos após AVC ou angioplastia coronariana. A perda da eficácia desse fármaco resulta em reestenose ou recidiva da trombose do vaso sanguíneo ou do stent, frequentemente com consequências graves. O clopidogrel é um profármaco metabolizado em sua forma ativa pelas enzimas P450, inclusive P450 2C19. Recentemente, polimorfismos da enzima P450 2C19 foram associados à redução do efeito antiplaquetário e aumento da morbidade cardiovascular. Além disso, como muitos inibidores da bomba de prótons também são metabolizados pela enzima P450 2C19, a administração concomitante de clopidogrel e um desses medicamentos de uso comum pode acarretar redução dos níveis plasmáticos de clopidogrel ativo.
Raça e etnia:
Alguns aspectos genéticos de raça e/ou etnia afetam o metabolismo dos fármacos. Em particular, as diferenças observadas nas ações de fármacos entre raças/etnias têm sido atribuídas a polimorfismos em genes específicos. Por exemplo, a P450 2D6 é funcionalmente inativa em 8% dos caucasianos, porém em apenas 1% dos asiáticos. Além disso, os afro-americanos exibem alta frequência de um alelo da P450 2D6, que codifica uma enzima com atividade diminuída. Essas observações são clinicamente relevantes, visto que a P450 2D6 é responsável pelo metabolismo oxidativo de cerca de 20% dos fármacos – incluindo muitos antagonistas β-adrenérgicos e antidepressivos tricíclicos – e pela conversão da codeína em morfina. Em alguns casos, um polimorfismo no gene-alvo constitui a base de diferenças étnicas observadas na ação de determinados fármacos. A atividade da enzima epóxido redutase de vitamina K (ERVK), alvo do anticoagulante varfarina, é afetada por polimorfismos de nucleotídios simples (PNS), que tornam um indivíduo mais ou menos sensível à varfarina e que determinam a administração de doses mais baixas ou mais altas do fármaco, respectivamente. Em um estudo, foi constatado que certas populações asiático-americanas apresentavam haplótipos (combinações herdadas de polimorfismos SNP) associados a sensibilidade aumentada à varfarina, enquanto populações afro-americanas exibiam haplótipos associados a aumento da resistência a tal substância. Talvez o exemplo mais proeminente de terapia baseada em polimorfismo genético seja a associação de dinitrato de isossorbida e hidralazina em dose fixa. Foi relatado que essa associação de vasodilatadores produz redução de 43% na taxa de mortalidade de afro-americanos com insuficiência cardíaca. Embora a base bioquímica desse efeito não seja conhecida, esses dados clínicos demonstram que os polimorfismos genéticos merecem consideração essencial na escolha do tratamento e das doses de um fármaco.
Idade e sexo:
O metabolismo dos fármacos também pode diferir entre indivíduos como resultado de diferenças de idade e sexo. Muitas reações de biotransformação são mais lentas tanto em crianças de pouca idade quanto em indivíduos idosos. Ao nascerem, os recém-nascidos são capazes de efetuar muitas das reações oxidativas, mas não todas elas; todavia, a maioria desses sistemas enzimáticos envolvidos no metabolismo de fármacos amadurece gradualmente no decorrer das primeiras 2 semanas de vida e durante toda a infância. É interessante lembrar que a icterícia neonatal resulta de uma deficiência da enzima de conjugação da bilirrubina, a UDPGT. Outro exemplo de deficiência de enzima de conjugação associada a risco de toxicidade em lactentes é a denominada síndrome cinzenta do recém-nascido. As infecções por Haemophilus influenzae em lactentes eram antigamente tratadas com o antibiótico cloranfenicol; a excreção desse fármaco exige transformação oxidativa, seguida de reação de conjugação. O metabólito de oxidação do cloranfenicol é tóxico; se esse metabólito não sofrer conjugação, seus níveis poderão aumentar no plasma, alcançando concentrações tóxicas. Como consequência da presença de níveis tóxicos do metabólito, os recém-nascidos podem sofrer choque e colapso circulatório, produzindo a palidez e a cianose que deram o nome a essa síndrome.
No indivíduo idoso, observa-se diminuição geral de sua capacidade metabólica. Consequentemente, é preciso um cuidado especial na prescrição de fármacos para esse segmento da população. O declínio da capacidade metabólica observado nos idosos tem sido atribuído à redução relacionada com a idade de massa hepática, fluxo sanguíneo hepático e, possivelmente, atividade das enzimas hepáticas. Outra consideração terapêutica é que os idosos frequentemente ingerem muitos medicamentos, com consequente aumento do risco de interações medicamentosas. Há algumas evidências de diferenças no metabolismo de fármacos em ambos os sexos, embora os mecanismos envolvidos não estejam bem elucidados, e os dados obtidos de animais de laboratório não sejam particularmente esclarecedores. Ocasionalmente, tem sido relatada diminuição na oxidação de etanol, estrógenos, benzodiazepínicos e salicilatos nas mulheres em comparação aos homens, a qual pode estar relacionada com os níveis de hormônios androgênicos.
Dieta e ambiente:
Tanto a dieta quanto o ambiente podem alterar o metabolismo dos fármacos ao induzir ou inibir as enzimas do sistema P450. Exemplo interessante é o suco de toranja (grapefruit). Os derivados do psoraleno e os flavonoides encontrados nesse suco inibem tanto a P450 3A4 quanto o MDR1 no intestino delgado. A inibição da enzima diminui significativamente o metabolismo de primeira passagem de fármacos coadministrados que também são metabolizados por essa enzima, enquanto a inibição do MDR1 aumenta de modo substancial a absorção de fármacos coadministrados que são substratos para efluxo por essa enzima. O efeito do suco de toranja é importante quando esse suco é ingerido com fármacos metabolizados por essas enzimas. Tais fármacos incluem alguns inibidores da protease, antibióticos macrolídeos, inibidores da hidroximetil glutaril CoA redutase (estatinas) e bloqueadores dos canais de cálcio. O saquinavir é um dos inibidores da protease metabolizado pela P450 3A4 e exportado pelo MDR1. No caso descrito no início deste capítulo, a Srta. B deveria ter sido alertada quanto ao fato de que a ingestão simultânea de suco de toranja e saquinavir pode resultar inadvertidamente em níveis séricos tóxicos do inibidor da protease.
Os fitoterápicos também exercem efeitos significativos no sistema P450. Um exemplo é a ervade-são-joão, fitoterápico popular usado para estabilização do humor. Muitos estudos observacionais constataram que o hipérico consegue induzir a expressão de P450 e, assim, reduzir a eficácia de outras substâncias. Componentes de plantas e condimentos também inibem P450. Um exemplo é a piperina (composto químico essencial da pimenta-do-reino), que comprovadamente inibe P450 3A4 e a proteína MDR em modelos animais. A importância clínica desse efeito ainda não foi determinada. Como muitas substâncias endógenas utilizadas nas reações de conjugação derivam, em última análise, da dieta (e também necessitam de energia para a produção dos cofatores apropriados), a nutrição pode afetar o metabolismo dos fármacos ao alterar o reservatório dessas substâncias disponíveis para as enzimas de conjugação. A exposição a poluentes pode, de modo semelhante, produzir efeitos radicais sobre o metabolismo dos fármacos; um exemplo é a indução das enzimas P450 mediada por AhR, ocasionada por hidrocarbonetos aromáticos policíclicos presentes na fumaça do cigarro.
Interações medicamentosas metabólicas:
Os fármacos potencialmente afetam biodisponibilidade oral, ligação às proteínas plasmáticas, metabolismo hepático e excreção renal de fármacos coadministrados. Entre as categorias de interações medicamentosas, os efeitos sobre a biotransformação têm importância clínica especial. O conceito de indução e inibição das enzimas P450 já foi introduzido. Situação clínica comum que precisa levar em consideração esse tipo de interação medicamentosa é a prescrição de determinados antibióticos a mulheres em uso de contracepção hormonal. Por exemplo, a indução enzimática pelo antibiótico rifampicina faz com que a contracepção hormonal à base de estrógeno seja ineficaz em doses convencionais, visto que a rifampicina induz a P450 3A4, que é a principal enzima envolvida no metabolismo do componente estrogênico comum, o 17α-etinilestradiol. Nessa situação, é necessário recomendar outros métodos de contracepção durante o tratamento com rifampicina. A Srta. B deve ser alertada dessa interação se a rifampicina for acrescentada ao esquema terapêutico. A erva-de-são-joão, um fitoterápico, também produz indução da P450 3A4, e, por conseguinte, apresenta efeito semelhante sobre a contracepção hormonal à base de estrógeno. Outro fenômeno associado à indução enzimática é a tolerância, que pode ocorrer quando um fármaco induz seu próprio metabolismo e, dessa maneira, diminui sua eficácia com o decorrer do tempo.
Como os fármacos são frequentemente prescritos em associação a outros produtos farmacêuticos, deve-se dispensar cuidadosa atenção aos metabolizados pelas mesmas enzimas hepáticas. A administração concomitante de dois ou mais fármacos metabolizados pela mesma enzima resultará, em geral, em níveis séricos mais elevados dos medicamentos. Os mecanismos de interação medicamentosa podem envolver inibição competitiva dos substratos, inibição alostérica ou inativação enzimática irreversível; em qualquer um dos casos, pode-se verificar elevação aguda dos níveis de fármacos, induzindo, possivelmente, resultados deletérios. Por exemplo, a eritromicina é metabolizada pela P450 3A4, porém o metabólito nitrosoalcano resultante pode formar um complexo com a P450 3A4 e inibir a enzima. Essa inibição pode levar a interações medicamentosas potencialmente fatais. Exemplo notável é a interação entre a eritromicina e a cisaprida, fármaco que estimula a motilidade do trato GI. As concentrações tóxicas de cisaprida podem inibir os canais de potássio HERG no coração e, assim, induzir arritmias cardíacas potencialmente fatais; por esse motivo, a cisaprida foi retirada do mercado em 2000. Antes de sua retirada, era frequentemente bem tolerada como agente único. Entretanto, como a cisaprida é metabolizada pela P450 3A4, quando a atividade dessa enzima é comprometida em decorrência da administração concomitante de eritromicina ou de outro inibidor enzimático, as concentrações séricas de cisaprida podem aumentar e alcançar níveis associados à indução de arritmias. Em outros casos, as interações medicamentosas podem ser benéficas. Por exemplo, conforme assinalado, a ingestão de metanol (componente do álcool metílico) pode resultar em cegueira ou morte, visto que seus metabólitos (formaldeído, agente utilizado no embalsamamento, e ácido fórmico, componente do veneno de formiga) são altamente tóxicos. Um tratamento para o envenenamento por metanol consiste na administração de etanol, que compete com o metanol pela oxidação mediada pela álcool desidrogenase (e, em menor grau, pela P450 2D1). A consequente demora na oxidação possibilita a depuração renal do metanol antes que possa haver formação de seus subprodutos tóxicos no fígado.
Doenças que afetam o metabolismo dos fármacos:
Muitos estados mórbidos podem afetar a velocidade e a extensão do metabolismo de fármacos no corpo. Como o fígado constitui o principal local de biotransformação, várias doenças hepáticas comprometem significativamente o metabolismo dos fármacos. Hepatite, cirrose, câncer, hemocromatose e esteatose hepática podem comprometer as enzimas do citocromo P450 e outras enzimas hepáticas cruciais para o metabolismo dos fármacos. Como consequência desse metabolismo mais lento, os níveis das formas ativas de muitos fármacos podem atingir valores mais altos do que o desejado, causando efeitos tóxicos. Por conseguinte, talvez seja necessário reduzir as doses de muitos fármacos em pacientes com hepatopatia. A doença cardíaca concomitante também pode afetar o metabolismo dos fármacos. A intensidade do metabolismo de muitos fármacos, como o antiarrítmico lidocaína e o opioide morfina, depende da ▶ liberação de fármacos no fígado por meio da corrente sanguínea. Como o fluxo sanguíneo está comumente comprometido na doença cardíaca, é preciso atentar para o potencial de níveis supraterapêuticos de fármacos em pacientes com insuficiência cardíaca. Além disso, alguns agentes anti-hipertensivos reduzem de modo seletivo o fluxo sanguíneo para o fígado e, assim, podem aumentar a meia-vida de um fármaco como a lidocaína, resultando em níveis potencialmente tóxicos. O hormônio tireoidiano regula o metabolismo basal do corpo, que, por sua vez, afeta o metabolismo dos fármacos. O hipertireoidismo pode aumentar a intensidade do metabolismo de alguns fármacos, enquanto o hipotireoidismo pode ter o efeito oposto. Acredita-se também que outras afecções, como doença pulmonar, disfunção endócrina e diabetes, afetem o metabolismo dos fármacos, porém os mecanismos envolvidos nesses efeitos ainda não estão totalmente elucidados.